
Abstract
An online survey was conducted by the International Life Sciences Institute, Food Biotechnology Committee, on the use of qualitative and quantitative polymerase chain reaction (PCR) assays for cauliflower mosaic virus 35S promoter and Agrobacterium tumefaciens Tnos DNA sequence elements for the detection of genetically engineered (GE) crop plant material. Forty-four testing laboratories around the world completed the survey. The results showed the widespread use of such methods, the multiplicity of published and in-house methods, and the variety of reference materials and calibrants in use. There was an interest on the part of respondents in validated quantitative assays relevant to all GE events that contain these two genetic elements. Data are presented by testing two variations each of five published real-time quantitative PCR methods for 35S detection on eight maize reference materials. The results showed that two of the five methods were not suitable for all the eight reference materials, with poor linear regression parameters and multiple PCR amplification products for some of the reference materials. This study demonstrates that not all 35S methods produce satisfactory results, emphasizing the need for method validation.
Similar content being viewed by others

Introduction
Vector constructs for plant transformation contain sequences of DNA which are intended to be inserted into the target organism [1]. In addition to the sequences, which may be required for insertion of the construct into the plant genome such as T DNA borders, a vector construct includes promoter and terminator sequences that enable the plant to express the gene of interest. One source of such promoters is the cauliflower mosaic virus (CaMV). CaMV is a double-stranded DNA virus affecting plants in the Cruciferae, Resedaceae, and Solanaceae [2]. The 35S promoter of CaMV is a functional, well-characterized, and constitutively expressed promoter [3]. Hence it has been incorporated into numerous constructs and used to produce many of the GE crops that are in commercial production, such as maize, soy, canola, and papaya. Similarly the RNA polyadenylation site (indicating the end of transcription) of the Tnos sequence from the Agrobacterium tumefaciens nopaline synthase gene has served as a polyadenylation site in some of the same constructs. The number of GE events and GE products in 17 different taxa containing either or both 35S promoter and Tnos sequences is summarized in Table 1. Not all of these are commercialized. Maize has the largest number of GE products at 27. In recent years, many of the maize events have been crossed using normal breeding techniques to produce what are called stacked-trait products. The Agbios database [4] shows that there are 19 such double- and triple-stacked maize products, and to our knowledge there are no stacked products that do not contain either 35S or Tnos sequences. The last column of Table 1 indicates how relatively few GE products have neither 35S nor Tnos sequences.
Testing for the presence of CaMV 35S and Tnos sequences has been commonly used as a screening tool for detection of GE plant material since most or all GE events/products in commerce contained one or the other or both. Detection of either element requires additional assays for identification of specific traits or events [5]. This involves the use of qualitative PCR assays with the products separated by gel or capillary electrophoresis. The use of multiplex assays and microarrays are recent developments [6, 7] and provide alternative methods to identify GE products. Finally, a quantitative assay based on the identified product can be used to quantify GE material in food or grain. This type of event-specific assay may target the junction between the transgene construct and the plant genomic DNA that is unique to any given event.
One approach to quantification that has been demonstrably successful in some diagnostic laboratories is the use of 35S or Tnos as the target sequence [8]. Since these DNA sequences are still common to most commercially grown crops, an internationally recognized and validated quantitative method would be useful and could, in some cases, substitute for the quantification of unique event assays. The method would have to be validated for all products carrying 35S and Tnos, if possible. The products for which quantitative screening assays are not appropriate would have to be clearly understood. With some products there is more than one copy of these elements, such as maize Bt11 that has two copies of the 35S sequence. This potentially leads to an overestimate of the GE content and could be problematic when the GE content is near a regulatory threshold.
Complex mixtures containing more than one GE product may be identified and could be due to the presence of a stacked-trait product, for example maize Mon 810 × Mon 88017, or to the independent presence of two distinct GE products. Currently there is no good analytical approach for distinguishing between these two possibilities. While the presence of equivalent quantities in a sample of both Mon 810 and Mon 88017, for example, could be due to the presence of the same quantity of two different products, this situation would be suggestive of the presence of a stacked-trait product. Depending on the stacked-trait product, quantification by 35S or Tnos may or may not involve extra copies per haploid genome of these elements. The stacked-trait product, Maize Mon 810 × GA21, has one copy each of 35S and Tnos elements/two haploid genomes with Mon810 containing the 35S and GA21 containing Tnos. Two event-specific assays would estimate twice the GE content compared with the 35S or Tnos assay alone. In the case of the stacked-trait maize Mon810 × Mon 88017 there are two 35S-containing transgene constructs, one from each parent, for an equivalent of one 35S copy per haploid genome. Quantification by 35S qPCR is used by the Japanese government in their testing of imported food and grain. There are several advantages to this strategy. One validated method could substitute in appropriate cases for event-specific quantitative assays. The cost of testing would be reduced, and the efficiency of testing could be increased by combining diverse test materials in a given assay run. Widespread adoption of such a method may lead to more consistency of testing of materials upon export and subsequent import, reducing the number of trade disputes. In addition, for those laboratories using a 35S assay only for qualitative purposes, switching to a real-time quantitative method would eliminate the need for post amplification processing, such as detection using agarose gel electrophoresis or capillary electrophoresis. Quantitation by 35S or Tnos elements could be useful tools, but knowledge of the products and regulatory requirements would be important for appropriate application.
The goal of this manuscript was first to determine which methodologies are currently used around the world to detect GE crops and then to assess the validity of the methods used. Accordingly, we describe the results of a survey conducted by the International Life Science Institute (ILSI) on the use of 35S and Tnos methods by the international testing community to determine the extent of use and interest in such methods. We then provide data on 35S measurements of eight maize products using five published methods. The data show that some 35S methods utilized in this study were not suitable for all the maize products, thus emphasizing the need to validate such a 35S detection method for each GE event.
International Life Science Institute survey on the use of the transgene elements 35S promoter and Tnos for detection of genetically engineered plant materials
To assess the status of 35S and Tnos PCR-based detection methods currently in use, ILSI petitioned over 100 laboratories with testing experience for GE traits, requesting participation in a survey. The scope of the survey was to collect information on the use of qualitative and quantitative PCR-based methods for 35S and Tnos by the laboratories. The survey was done online using Survey Monkey [9]. Twenty-five questions were asked about current and past use, the type of methods and detection strategies, the source of methods (published versus in-house), and types of reference materials. Each participant in the survey was allowed one survey submission.
There were 46 separate accessions to the survey, and 44 of these laboratories completed the survey. Identification was not obligatory, though 26 were willing to be identified in a participant list. Thirty-two of the participants identified at least their country. The geographic distribution of the laboratories is as follows: 14 from Europe (Germany, Poland, Portugal, France, and Spain), ten from North America (Canada, USA, and Mexico), three from South America (Argentina, Nicaragua, and Brazil), and five from Asia (China, Thailand, and India). The countries with the most respondents included Germany and the USA, both with seven.
Of the 44 laboratories, 40 currently use a qualitative only, a quantitative only, or a combination of qualitative and quantitative PCR assays for 35S, while 37 use some combination of assays for Tnos. Similar numbers were seen when the question of past usage was asked. The use of qualitative methods has dropped from 37 to 33 labs for 35S and from 34 to 32 labs for Tnos. Correspondingly, the current use of quantitative assays increased from past use: from 19 to 22 labs for 35S and from 12 to 16 for Tnos. The laboratories that do not currently use quantitative assays for 35S and Tnos were asked if they were considering using a quantitative assay; eight of 22 respondents said yes and 14 of 22 said no for 35S, while eight of 25 said yes for Tnos and 17 of 25 replied in the negative.
Next the laboratories were queried as to the type and source of their qualitative methods. All laboratories reported that qualitative methods for detection of both 35S and Tnos elements are PCR methods. Published methods for 35S were used by 24 of 38 respondents (63%), while 14 of 38 (37%) of the laboratories used methods developed in-house. For Tnos, 13 of 35 (37%) laboratories used in-house developed methods, while 22 of 35 laboratories use published methods. For qualitative assays, detection of the PCR product is done using agarose gel electrophoresis by 66% (23 of 35) of respondents for both 35S and Tnos assays. The other 12 laboratories (34%) use other techniques, such as TaqMan real-time PCR (seven labs), the Agilent 2100 Bioanalyzer, SYBRGreen, and polyacrylamide gel electrophoresis.
All of the laboratories that reported performing a quantitative assay for 35S or Tnos use real-time PCR; 13 out of 25 use published 35S methods, and eight out of 17 use published Tnos methods. For quantitative assays, 85% of the respondents (19 labs) use probes labeled with fluorophores, while the remainder used fluorescent intercalating dyes such as SYBRGreen.
The survey respondents were queried as to the source of the published methods for both qualitative and quantitative methods, and Table 2 summarizes what was provided in the survey. There were some specific journal references provided, but in some cases sources were general such as the Joint Research Center GMO database and GMDD [10, 11]. The survey respondents did not specify which method(s) within the databases was used. Table 2 shows the variety of unique references for each of the four categories of methods found in the named sources. Some of the references are redundant in the sense that they appear in more than one source.
The respondents were asked to identify what endogenous control gene they use when doing relative quantification. Twenty-one laboratories identified gene targets in four taxa. For maize, five targets were specified: alcohol dehydrogenase, invertase, high mobility group, starch synthase (SSIIb), and zein. For rape (canola), laboratories use cruciferin, fatty acid dehydrogenase, or phosphoenolpyruvate carboxylase. One target, phospholipase, was indicated for rice and lectin was indicated for soy. When considering the variety of endogenous control gene targets, there is likely even more variety in possible qPCR assays since more than one primer/probe system exists for some of these target sequences, and the reaction kinetics are not necessarily equivalent [12]. One respondent stated that they use the endogenous control gene assays provided on the Community Research Laboratories web site [13]. Another laboratory indicated that they use a chloroplast gene, but did not specify which taxa were relevant for use of that target or how it was used.
The next section of the survey concerned the use of reference materials (RM). The participants were initially asked if they use commercial RMs for calibration and quality control. Of 35 total responses, 27 said that they use commercial RMs for calibration of 35S assays and 26 use such materials for quality control. Twenty-two labs use commercial RMs for Tnos calibration and 23 for quality control. In a related question, 25 of 30 labs reported using certified reference materials (CRM) from sources such as the Institute for Reference Materials and Measurements (IRMM) [14] and the American Oil Chemists Society (AOCS) [15]. Eleven respondents indicated that they used other materials for calibration. These included in-house developed RMs, plasmids, seeds, and materials from proficiency testing programs and inter-laboratory trials. Then, respondents were asked to indicate what categories of materials they use (Table 3). Many respondents indicated use of more than one type of material, but the most popular type is powder, such as CRMs produced by IRMM. When asked if there were additional comments on reference materials, seven participants noted the lack of reference materials for some of the GE products of interest, including 35S and Tnos. In some cases, laboratories expressed frustration in obtaining the commercial RMs. There was also concern about the cost of RMs and the shelf life of an opened vial of a CRM. One comment stated a preference for powder materials, as this required extraction (unlike pure DNA RMs), thus covering the whole process of DNA extraction and PCR assay.
The survey participants were asked if their qualitative and quantitative 35S and Tnos assays were able to detect all the events that the participants encounter in their testing. Twelve participants replied yes and 20 said no for their 35S assays, while 12 said yes and 17 no for Tnos assays. Some respondents mentioned that they test for events that do not contain either of the two targets in the genome. Others suggested that the assays work for all the events that they test for, and several pointed out the necessity for using both assays. One laboratory noted low levels and adventitious presence of test samples with “spurious Roundup ready soy.” Precautions noted by the respondents include the importance of confirmation using event or construct specific methods and controlling for false positives due to the presence of actual cauliflower mosaic virus (the original source of the 35S promoter). There are PCR methods available that target other regions of the cauliflower mosaic virus that can serve as control assays for virus contamination [16, 17]. One lab noted that not all primer/probe combinations will successfully amplify all events, but did not indicate if that referred to 35S or Tnos assays or both. The copy number of these elements in specific events was also of concern to some of the respondents with respect to quantitative assays. One lab noted that they thought the assays had low sensitivity and reproducibility.
Perhaps the most interesting aspect of the survey is that there is interest in adopting a standardized method, if available. Of 32 respondents, 16 were “highly interested,” 13 were “somewhat interested,” and three were not interested in a standardized 35S method. The equivalent numbers for Tnos were 16, 13, and 4. Some respondents were of the opinion that there were already sufficiently standardized methods available. Some noted that a standardized method would be an improvement and could lead to better inter-laboratory reproducibility. Possible problems noted by participants could be the regulatory requirements in specific countries, the flexibility of a standardized method such as core reagent selection, and the cost of validating a new method in-house by a lab investigating adoption of the standardized method.
It is clear from this survey that a plethora of 35S and Tnos methods exists along with a variety of reference materials for calibration and quality control purposes that lack standardization in laboratories globally. Such heterogeneity in methodology can potentially cause problems for the food/grain production and trade industry due to the global nature of trade. Use of internationally recognized standardized methods by laboratories at export and import sites could reduce the possibility of trade disputes.
Method screening experiments
Based on the survey results, we conducted preliminary experiments to assess and compare the performance of five 35S qPCR methods that are publicly available in the literature. The test materials were eight different maize CRMs.
Materials and methods
Testing materials for this study consisted of certified reference materials from IRMM and AOCS. IRMM has produced CRMs for many GE crops. The IRMM CRMs [14] included in this study are matrix materials, ground maize seed, with a percentage of GE material in a background of isogenic conventional corn up to ~ 10% (100 g/kg, W/W). We used seven of these CRMs in this study at the highest concentration available for a specified event. The eighth material was a pure DNA preparation isolated from leaves of homozygous transgenic T25 maize and thus was 100% GE material. The T25 material was certified by AOCS [15]. Table 4 identifies the specific CRMs used in this study.
DNA was extracted from the seven matrix materials using a publicly available CTAB method validated for maize TC1507 [18]. The method calls for two different cleanup steps. In this study, only one step was used, the S-300 HR Microspin columns (Amersham-Pharmacia)Footnote 1, since the second cleanup step resulted in DNA absorbance scans that were of poorer quality (smaller 260:280 nm ratios), suggesting that impurities were introduced. Extractions were done with 100 mg maize flour. Seven to eight extractions were done from each material and were pooled after performing a wavelength scan of each individual extract. The 260:280 nm ratio ranged from 1.92 to 1.97, and the 260:230 nm ratio was always above 2.0 for all samples. The few DNA samples that did not meet these criteria were discarded. The absorbance at 260 nm was measured with the DNA in 0.2× TE buffer and after the addition of 2 M NaOH. The calculation of the alkali denatured DNA concentration (μg mL−1) was on average 11% lower than DNA in buffer. The alkali denatured DNA value was used in subsequent calculations, and the DNA was adjusted to approximately 20 μg mL−1 (17.5 to 22.0 μg mL−1), except for the T25 DNA, which was adjusted to 1 μg mL−1. This adjustment was done to bring the GE DNA copy number for the PCR assays into the same range for all materials. The extracted DNA was size separated using agarose gel electrophoresis (0.8% agarose, constant voltage of 100 V, 1.5 h) and stained with ethidium bromide. All DNA was observed to be intact with minimal degradation, with the observed band in the range of 25,000 to 50,000 base pairs (bp).
From the concentration of DNA, the number of copies per assay was calculated using the 1C value (haploid genome mass in pg) derived from several references. The estimates from four references ranged from 2.57 to 2.8 pg per haploid genome [19–21]. In our calculations, one haploid genome was considered to be approximately 2.6 pg, and each ng of maize DNA was equivalent to 385 haploid genome copies. The calculation of copies/ng for the DNA of any specific event took into consideration the mass fraction of GE corn in the CRM and the zygosity of the materials. T25 DNA was purified from the leaves of the inbred line and thus is homozygous (one copy per haploid genome). Bt11 has two copies of 35S in its transgene construct, making it equivalent to the homozygous T25. The matrix maize CRMs are hybrid seed. Therefore, except for Bt11, there is on average one copy of the transgene construct per two haploid genomes (or 0.5 copies/haploid genome). This is the hemizygous state. Maize seed tissue is composed of diploid embryo and triploid endosperm with the female parent donating the third set of chromosomes to the endosperm tissue; thus, the actual fractional copy number can range from 0.4 to 0.7 copies/haploid genome. This latter issue (triploid endosperm) was not taken into account in experiments reported here. We used 0.5 copies/haploid genome in our calculations of copy number of transgenes in the materials tested except for samples T25 and Bt11.
Five published quantitative real-time PCR methods [22–26] for amplification of the 35S element were selected and labeled as method 1 to method 5 for purposes of this study. All of these methods were selected because of the use of TaqMan probe technology and the small size of the amplicons, which ranged from 68 to 101 bp. All methods referenced the use of Applied Biosystem real-time PCR platforms, but not all the same model number. All but one used ABI Taqman® Universal PCR master mix. The cycling parameters described in each method were as recommended by the manufacturer for that type of assay and master mix. There were a few modifications to the cycling parameters in two methods: a shorter extension time in one method (30 versus 60 s, method 2) and a longer denaturation time in the second method (30 versus 15 s, method 1). The locations of the primers on the sequence of the CaMV 35S promoter sequence, for the five methods, are identified in Fig. 1. The entire promoter sequence is not shown, only the sequence data relevant to this study.

Quantitative real-time PCR assays were conducted at the United States Department of Agriculture-Grain Inspection, Packers and Stockyards Administration (USDA-GIPSA) and the National Institute of Standards and Technology (NIST). Assays conducted in the USDA-GIPSA laboratory were as described in the published methods [22–26]. Assays were run on an ABI 7900 instrument using Taqman® Universal PCR master mix at 1× final concentration and the following standard cycling parameters recommended for the ABI universal master mix: 2 min at 50 °C (UNG activation), 95 °C for 10 min (activation of Taq DNA polymerase), followed by 45 cycles of 95 °C for 15 s (denaturation), and 60 °C for 60 s (annealing/extension). A series of four 1:2 dilutions of the DNA were made from the ~20 μg/mL stock DNA, and 5 μL of DNA was added to the reaction mix. Each of the five DNA concentrations per product was assayed in triplicate, and the log transform of the copy number was plotted against the Ct value, and the linear regression curve parameters were calculated. The number of genome copies in the assays ranged from 60 to 2,000.
The NIST laboratory conducted assays on an ABI Prism 7000 with the primers at the recommended concentration but used SYBRGreen intercalating dye as the fluorescent detection agent (SYBRGreener Universal master mix for the ABI Prism, Invitrogen). NIST and USDA-GIPSA used the same preparation of primers. In the SYBRGreen assays the concentration of the primers was the same as described in the published methods. The assays were conducted with the following cycling parameters recommended for the master mix: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of annealing and extension at 95 °C for 15 s and 60 °C for 60 s. At the end of 40 cycles a melting curve analysis was performed.
The complete experimental procedure was conducted twice with respect to DNA extraction, DNA characterization, and SYBRGreen assays with very similar results. Data are not shown for the first set of extractions. Extra extractions were performed on TC1507 maize CRM material along with additional assays. The TaqMan assays were performed once at USDA-GIPSA on the second complete set of DNA extractions.
Experimental results
Experimental data were produced using five published quantitative real-time PCR methods for 35S DNA sequences to evaluate method variability on the eight GE maize CRMs. Data were not generated for Tnos sequences in this study. Since we utilized CRMs for this work with certified mass fractions of GE maize, we could compute the copy number of genomes containing the transgene construct. We made a series of dilutions of each of the extracted DNAs and assayed each of the five dilutions in triplicate. We then plotted the data (Ct value versus the log of the transgene copy number) and calculated a linear regression. The slope of the linear regression provided an evaluation of the efficiency of amplification with a slope −3.32 being ideal and equivalent to an efficiency of 100% [27, 28]. The correlation coefficient (R 2) indicated how closely the data points approximate the regression line. The Y-intercept indicates the Ct value that would be expected, if the amplification started with a single copy of a transgene construct present in the reaction mix.
The Y-intercept, slope, and R 2 data for the TaqMan assays are summarized in Table 5. The average Ct values for the extracts containing the highest and lowest DNA concentration were also determined. This was an easy way to compare the data from one method to another for a given DNA extract. The mean of the slopes across the events were calculated and ranged from −3.16 to −3.62, with an exception, TC1507 (methods 2 and 5), as discussed below. Only one curve had a correlation coefficient below 0.95 and most were 0.98 and above. The copy number for the GE product was calculated based on the quantity of DNA in maize sample, the mass fraction of event DNA, the zygosity, and copy number in the genome. Events 176, Mon810, and NK603 had about half as many copies per assay as did the equivalent DNA quantity for TC1507, Mon 863, and 59122 because of mass fraction differences of the certified RM (~5% versus ~10%). Bt11 was present at the 5% level in the certified RM but it has two copies of 35S per transgene construct. T25 (100% transgene) DNA was diluted to be equivalent in copy number to TC1507, Mon863, 59122, and Bt11. Therefore lower Ct values (~1 Ct) with those samples with the 2× higher level copy number for 35S were predicted. On average the Ct values of TC1507 (methods 1, 3, and 4 only), Mon 863, 59122, and T25 tended to be lower (28.2, 28.6, 29.1, and 28.5, respectively) than Bt176, Mon810, and NK603 (29.6, 29.6, and 29.5, respectively). Note that the Bt11 data were not consistent. No statistically significant difference is claimed since insufficient data were available for statistical analysis, but the trend was generally what was predicted.
The Y-intercept, slope, and R 2 data for the SYBRGreen assays are summarized in Table 6. The data are plotted as for the TaqMan assays with the addition of a melting curve analysis. The Y-intercepts for the SYBRGreen assays were at a lower Ct value compared with TaqMan assays. Consistent with a lower Ct value for the Y-intercept were lower Ct values for the highest and the lowest DNA concentrations. The slopes were shallower on average than those of the TaqMan assays with the overall average slope equal to −3.16 for SYBR Green assays compared with −3.36 for the TaqMan assays. The range for the averaged SYBRGreen slopes was −2.80 to −3.45, not including maize TC1507.
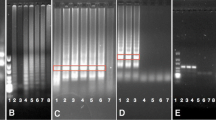
The SYBRGreen assays were a modification of the original published TaqMan method. The primary use of this modified method is to ascertain whether or not a single amplified product is produced in the assay as indicated by the presence of one peak (one PCR product) in the melting curve analysis. This analysis revealed that the only (or major) peak had the same melting temperature for each method across the eight event DNAs (Table 6). However, there were additional peaks seen in some assays (Table 6). Figure 2a, b shows melting curves for Bt11 assays in which a single peak was seen with the method 4 assays, while three peaks were seen with method 5, suggesting that multiple PCR products formed.

Melting curves for SYBRGreen assays of Bt11 showing a single peak for method 4 assays (a) and three peaks for method 5 assays (b)
The biggest anomaly was observed with maize TC1507 assayed by methods 2 and 5; see Tables 5 and 6 and Fig. 3. The SYBRGreen assays for methods 2 and 5 showed a very shallow slope, much larger Ct values (low and high concentrations), and very poor correlation coefficients. In addition, for method 5, evidence of two products was ascertained with the observation of a second peak characterized by a lower melting temperature (70 °C). The TaqMan assays for methods 2 and 5 with TC1507 also showed larger Ct values for the high and low DNA concentrations as compared to the other three methods (see Table 5). The slopes of TaqMan assays were less extreme than the SYBRGreen assays (−3.156 for TM versus −1.12 for SG and −2.729 for TM versus −2.01 for SG methods 2 and 5, respectively).

Plot of SYBRGreen assays of TC1507 using methods 1 and 2 with linear regressions. Assay parameters for method 1: R 2 = 0.982 and slope of −2.94; method 2: R 2 = 0.74 and slope of −1.12
Additional anomalies (methods 2 and 5) included multiple products seen when SYBRGreen assays were run on Bt11 DNA using method 5. A total of three peaks were observed, the expected one and two others (Fig. 2). The TaqMan assay on Bt11 using method 5 gave Ct values that were about Ct value later than methods one to 4. The SYBRGreen assays for Bt11 using method 2 showed a small shoulder on the peak that is the expected product. SYBRGreen assays on NK603 with method 5 also showed an additional product with a higher melting temperature than the peak of the expected product.
Discussion
The survey showed that a large variety of methods are in use for qualitative and quantitative detection of the 35S and Tnos elements. While a number of sources were cited for published methods, a significant percentage of laboratories (37% to 53%) are using in-house developed PCR assays for these genetic elements. Some laboratories are using real-time TaqMan assays for 35S and Tnos as a qualitative tool. A variety of calibrants and quality control materials, including CRMs, plasmids, and proficiency testing samples, are in use. The survey showed that there is interest in standardized methods for 35S and Tnos.
The survey responses led us to do a screening of some published 35S quantitative PCR methods using maize CRMs as test material. Amplification of different regions of the 35S promoter, as defined by the primer binding sites, and two different fluorescent detection strategies, TaqMan and SYBRGreen, were used for 35S detection. Methods 1, 3, and 4 provided consistent results with all GE certified reference materials. The primer binding sites for these three methods are in the same region of the 35S promoter element and produce amplicons related in sequence (see Fig. 1). Methods 1 and 4 share the same reverse primer and have overlapping forward primers. The results suggested that the region covered by these three methods is conserved in the DNA sequences of at least these eight GE products.
The data also revealed that not all 35S PCR methods are likely to give accurate quantitative results with all the GE products tested. Problems associated with methods 2 and 5 include multiple amplicons (PCR products) in some assays and inefficient PCR, a poor correlation coefficient, and larger Ct values with TC1507 maize. Similar observations were made with both TaqMan and SYBRGreen technology. Possible explanations for this observation include DNA inhibitors or sequence heterogeneity in primer binding sites. Additional extractions of TC1507 DNA and repetition of the assays showed the same result, making DNA inhibition a less likely explanation for the results. After this manuscript was submitted to the journal for review, a paper by Morisett et al. [29] was published that demonstrated a single nucleotide polymorphism (SNP) in the 35S promoter sequence of TC1507 maize. This SNP is located in the four base pair region where the forward primer for method 2 overlaps the reverse primer for method 5 (see Fig. 1, first line of the sequence for the region.) Such heterogeneity in the primer binding sites could result in inefficient amplification of TC1507 DNA giving rise to the results seen with methods 2 and 5.
The ILSI online survey showed that among the 44 laboratories that accessed and completed the survey, 33/32 use a qualitative 35S/Tnos assay and 22/16 use a quantitative 35S/Tnos assay. Fifty percent of the labs included in the survey are using a quantitative 35S method, but the number of laboratories using this as a quantification tool could not be ascertained in this survey. There appears to be continuing interest in using 35S and Tnos as targets for amplification. Table 1 shows that a large percentage of GE products in the Agbios GM Database have either or both 35S and Tnos sequences in their transgene constructs. Maize has the largest number of GE events by far (27), and it is increasingly common for farmers to plant maize seed that are stacked-trait products. There are 18 of these in the database. This increases the likelihood that 35S or Tnos sequences are going to be in the genome of the harvested grain. While some recent products and others under development have transgene constructs that use alternative promoter and terminator sequences, 35S and Tnos are likely to remain an important component of commercial GE food and feed products for the foreseeable future.
The laboratories in the survey perform a variety of PCR assays, some of which are developed in-house, while others came from the literature, ISO standards, databases, and official sources (Table 2). Some methods have gone through a validation process and inter-laboratory studies [24, 30]. There is no method in the literature that has been checked with all the multiplicity of events that are available in commerce. Alterations, often proprietary, to the 35S and Tnos sequences made during the construction of the promoter-gene-poly A site junctions can be the source of error, as the primers may be targeted to sequences that do not exist or are altered in the DNA construct, rendering a given assay non-functional. In this study a SNP in TC1507in primer binding sites for two of the methods had a severe affect on amplification.
Quality measurements depend on the use of validated methods, determination of the measurement uncertainty, and the availability of reference materials as well as components including appropriate calibrated equipment, trained operators, quality reagents, and the quality of the extracted DNA. Inter-laboratory variability is exacerbated by the use of multiple methods, and limited availability of reference materials has ramifications for world trade in food and grain possibly leading to trade disputes. The use of standardized internationally recognized methods for 35S and Tnos assays, shown to work with all events/products in commerce, would make a significant contribution to international trade harmonization.
Notes
Certain commercial equipment and materials are identified to specify the experimental procedure. This does not imply recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the material or equipment is the best available for the purpose
References
Lee L-Y, Gelvin SB (2008) T-DNA binary vectors and systems. Plant Physiol 146:325–332
Plant Viruses Online. http://image.fs.uidaho.edu/vide/refs.htm. Accessed March 2009
Benfey PN, Chua N-H (1990) The cauliflower mosaic virus 35S promoter: combinatorial regulation of transcription in plants. Science 250:959–966
Agbios GM Database. http://www.agbios.com/main.php. Accessed June 4, 2009
Lipp M, Shillito R, Giroux R, Spiegelhalter F, Charlton S, Pinero D, Song P (2005) Polymerase chain reaction technology as analytical tool in agricultural biotechnology. J AOAC Int 88:136–155
Chaouachi M, Chupeau G, Berapd A, McKhann H, Romaniuk M, Giancola S, Daval V, Bertheau Y, Brunel D (2008) J Agric Food Chem 56:11596–11606
Leimanis S, Hamels S, Naze F, Mbella MM, Sneyers M, Hochegger R, Broll H, Roth L, Dallmann K, Micsinal A, LaPaz JL, Pla M, Brunen-Nieweler C, Papazova N, Taverniers I, Hess N, Kirschneit B, Bertheau Y, Audeon C, Laval V, Busch U, Pecoraro S, Neumann K, Rosel S, VanDijk J (2008) Kok Esther, Bellocchi G, Foti N, Mazzara M, Moens W, Remacle J, Van Den Eede G. Eur Food Res Technol 227:1621–1632
Layton DT, Spieglehalter F, Jenkins R (2008) How grain companies are managing the challenges posed by stacked events in meeting the global regulatory and commercial requirements for non-GM corn shipments: a comparison of methods in current use. 1st Global Conference on GMO Analysis, Como Italy. http://gmoglobalconference.jrc.ec.europa.eu/2008/Posters/T.1.22%20Layton_Como%20poster%20061608.pdf
Survey Monkey. http://www.surveymonkey.com. Accessed June 22, 2009
European Commission, Joint Research Center, GMO methods database. http://mbg.jrc.ec.europa.eu/home/ict/methodsdatabase.htm. Accessed June 22, 2009
Dong W, Yang L, Shen K, Banghyun K, Kleter GA, Marvin HJP, Guo R, Liang W, Zhang D (2008) GMDD: a database of GMO detection methods. BMC Bioinformatics 9:260 http://gmdd.shgmo.org/, accessed June 22, 2009
Scholdberg TA, Norden TD, Nelson DD, Jenkins GR (2009) Evaluating precision and accuracy when quantifying different endogenous control reference genes in maize using real-time PCR. J Agric Food Chem 57:2903–2911
European Commission, Joint Research Center, Community reference laboratory. http://gmo-crl.jrc.ec.europa.eu/. Accessed June 22, 2009
European Commission, Joint Research Center, Institute for Reference Materials and Measurements. http://irmm.jrc.ec.europa.eu/html/homepage.htm. Accessed June 22, 2009.
American Oil Chemists Society. www.aocs.org
Wolf C, Scherzinger M, Wruz A, Pauli U, Hubner P, Luthy J (2000) Detection of cauliflower mosaic virus by the polymerase chain reaction: testing of food components for false positive 35S-promoter screening results. Eur Food Res Technol 210:367–372
Chaouachi M, Fortabat MN, Geldreich A, Yot P, Kerlan C, Kevdani N, Audeon C, Romaniak M, Bertheau Y (2008) An accurate real-time PCR test for the detection and quantification of cauliflower mosaic virus (CaMV): applicable in GMO screening. Eur Food Res Technol 227:789–798
EC Joint Research Center, Community Reference Laboratory, DNA Extraction method. http://gmo-crl.jrc.ec.europa.eu/summaries/TC1507-DNAextrc.pdf. Accessed May 12, 2009
Arumuganathan K, Earle ED (1991) Nuclear DNA content of some important plant species. J Agric Food Chem 9:208–218
Rayburn AL, Biradar DP, Bullock DG, McMurphy LM (1992) Nuclear DNA content in F1 hybrids of maize. Heredity 70:294–300
Bennett MD, Leitch IJ (2004) Plant DNA C-values database (release 5.0, Dec. 2004). http://www.kew.org/cvalues/homepage.html. Accessed June 24, 2009
Kuribara H, Shindo Y, Matsuoka T, Takubo K, Futo S, Aoki N, Hirao T, Akiyama H, Goda Y, Toyada M, Hino A (2002) Novel reference molecules for quantitation of genetically modified maize and soybean. J AOAC Int 85:1077–1089 Method 1
Corbisier P, Trapmann S, Gancberg D, Hannes L, Van Iwaarden P, Berben G, Schimmel H, Emons H (2005) Quantitative determination of Roundup Ready soybean (Glycine max) extracted from highly processed flour. Anal Bioanal Chem 383:282–290 Method 2
Fernandez S, Charles-Delobel C, Geldreich A, Berthier G, Boyer F, Collonnier C, Coue-Philippe G, Diolez A, Duplan M-N, Kebdani N, Romaniuk M, Feinberg M, Bertheau Y (2005) Quantification of the 35S promoter in DNA extracts from genetically modified organisms using real-time polymerase chain reaction and specificity assessment on various genetically modified organisms. Part 1: operating procedure. J AOAC Int 88:5547–5557 Method 3
Zeitler R, Pietsch K, Waiblinger H-U (2002) Validation of real-time PCR methods for quantification of transgenic contamination in rape seed. Eur Food Res Technol 214:346–351 Method 4
Hohne M, Santisi CR, Meyer R (2002) Real-time multiplex PCR: an accurate method for the detection and quantification of 35S-CaMV promoter in genetically modified maize-containing food. Eur Food Res Technol 215:59–64 Method 5
Pfaffl MW (2004) In: Bustin SA (ed) A-Z of quantitative PCR. International University Line, La Jolla, pp 87–112
Brisson M, Hall S, Hamby RK, Park R, Srere HK (2004) In: Bustin SA (ed) A-Z of quantitative PCR. International University Line, La Jolla, pp 619–642
Morisett D, Demsar T, Gruden K, Vojvoda J, Stebih D, Zel J (2009) Detection of genetically modified organisms—closing the gaps. Nature Biotechnol 27:700–701
Feinberg M, Fernandez S, Cassard S, Charles-Delobel C, Bertheau Y (2005) Quantitation of 35S promoter in maize DNA extracts from genetically modified organisms using real-time polymerase chain reaction, part 2: interlaboratory study. J AOAC Int 88:558–573
Acknowledgments
We wish to thank the 44 laboratories who completed the questionnaire. We acknowledge the fine technical support at the NIST of Wade Reimonenq of Southern University A&M College, Baton Rouge, LA.
About ILSI
The ILSI is a nonprofit, worldwide foundation established in 1978 to advance the understanding of scientific issues relating to nutrition, food safety, toxicology, risk assessment, and the environment. ILSI also works to provide the science base for global harmonization in these areas. By bringing together scientists from academia, government, industry, and the public sector, ILSI seeks a balanced approach to solving problems of common concern for the well-being of the general public.
ILSI is headquartered in Washington, DC. ILSI branches include Argentina, Brazil, Europe, India, Japan, Korea, Mexico, North Africa and Gulf Region, North America, North Andean, South Africa, South Andean, Southeast Asia Region, the Focal Point in China, and the ILSI Health and Environmental Sciences Institute. ILSI also accomplishes its work through the ILSI Research Foundation. ILSI receives financial support from industry, government, and foundations.
Support
The survey described in this manuscript was supported by the members of the ILSI International Food Biotechnology Committee: BASF; Bayer CropScience; Cargill Inc., Dow AgroSciences; Monsanto Company; Masterfoods, Inc., Pioneer, A Dupont Company; and Procter & Gamble Company, Inc., Syngenta Biotechnology, Inc. CropLife International provided support for this project through an agreement with NIST and GIPSA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Holden, M.J., Levine, M., Scholdberg, T. et al. The use of 35S and Tnos expression elements in the measurement of genetically engineered plant materials. Anal Bioanal Chem 396, 2175–2187 (2010). https://doi.org/10.1007/s00216-009-3186-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-009-3186-x