
Abstract.
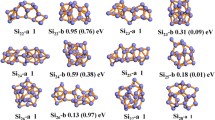
By an application to small silicon clusters Si N (with N = 4,5,7,10) it is shown that truly global geometry optimization on an ab initio or density functional theory level can be achieved, at a computational cost of approximately 1–5 traditional local optimization runs (depending on cluster size). This extends global optimization from the limited area of empirical potentials into the realm of ab initio quantum chemistry.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 24 February 1998 / Accepted: 6 March 1998 / Published online: 17 June 1998
Rights and permissions
About this article
Cite this article
Hartke, B. Global geometry optimization of small silicon clusters at the level of density functional theory. Theor Chem Acc 99, 241–247 (1998). https://doi.org/10.1007/s002140050332
Issue Date:
DOI: https://doi.org/10.1007/s002140050332