
Abstract
Biopharmaceuticals, monoclonal antibody (mAb)-based therapeutics in particular, have positively impacted millions of lives. MAbs and related therapeutics are highly desirable from a biopharmaceutical perspective as they are highly target specific and well tolerated within the human system. Nevertheless, several mAbs have been discontinued or withdrawn based either on their inability to demonstrate efficacy and/or due to adverse effects. Approved monoclonal antibodies and derived therapeutics have been associated with adverse effects such as immunogenicity, cytokine release syndrome, progressive multifocal leukoencephalopathy, intravascular haemolysis, cardiac arrhythmias, abnormal liver function, gastrointestinal perforation, bronchospasm, intraocular inflammation, urticaria, nephritis, neuropathy, birth defects, fever and cough to name a few. The advances made in this field are also impeded by a lack of progress in bioprocess development strategies as well as increasing costs owing to attrition, wherein the lack of efficacy and safety accounts for nearly 60 % of all factors contributing to attrition. This reiterates the need for smarter preclinical development using quality by design-based approaches encompassing carefully designed predictive models during early stages of drug development. Different in vitro and in silico methods are extensively used for predicting biological activity as well as toxicity during small molecule drug development; however, their full potential has not been utilized for biological drug development. The scope of in vitro and in silico tools in early developmental stages of monoclonal antibody-based therapeutics production and how it contributes to lower attrition rates leading to faster development of potential drug candidates has been evaluated. The applicability of computational toxicology approaches in this context as well as the pitfalls and promises of extending such techniques to biopharmaceutical development has been highlighted.
Similar content being viewed by others

Introduction
The pharmaceutical industry is currently valued at $786 billion from the total worldwide sales of prescription as well as over the counter drugs in 2015 wherein 25 % of this revenue was generated by biological/biotechnological products (Pharma 2014). Biological drugs are associated with living entities (cells and tissues) and/or their product such as recombinant therapeutic proteins and vaccines to name a few. Based on historical data, a shift towards biologics seems imminent owing to increasing profits and lower attrition rates when compared to small molecule drugs. Biological drugs comprised 70 % of the top ten selling products of the world in 2014, and the percentage sales of biotechnology products within the top 100 was 44 %. Twenty new biologicals were approved by FDA in 2014 compared to the 11 that were approved in 2009. Monoclonal antibodies have higher approval rates of 26 % in the biopharmaceutical sector than that of conventional small molecule drugs (10 %) (Hay et al. 2014). Based on the area of therapy, the largest segments of oncology and anti-rheumatoid drugs, which contribute to a combined compound annual growth rate of 13 %, continue to be dominated by biological drugs.
Even though the therapeutic efficiency of immunoglobulin molecules was demonstrated in 1890, it was only after Kohler and Milstein elucidated the murine hybridoma technology for in vitro production of mAbs (see Fig. 1 for generic mAb structures) that the market for mAbs grew and expanded to different therapy areas, such as haematology, oncology, immunology, cardiology, infectiology and ophthalmology as well as diagnostics and imaging(Köhler and Milstein 1975). The shift from murine mAbs to chimeric (human Fc region with murine Fv region) was mainly to increase titres as well as decrease immunogenic effects (Zhu 2012). To further decrease the murine composition and enhance Fc functionality, humanized mAbs were first developed in 1986 (Jones et al. 1985). The production systems routinely used for chimeric and humanized mAbs are Chinese hamster ovary (CHO) cells, NS0 and Sp2/0 myeloma cell line. To fully eliminate the immunogenic potential of murine epitopes while maintaining optimal Fc region functionality, fully human mAbs were developed by phage display technology and commercially produced by CHO system (Lai et al. 2013). Human embryonic kidney (HEK) and human retinal cell-derived (Per.C6) cell lines are the new potential candidates for biopharmaceutical production (Zhu 2012). In addition to being stable and producing high titres, the fully human cell lines offer the advantage of proper post-translation modification and glycosylation as they incorporate human biosynthetic pathways. Plant expression systems, such as recombinant Agrobacterium tumefaciens, and microbial systems, such as Escherichia coli, are gaining popularity for production of monoclonal antibodies against viruses (Berlec and Štrukelj 2013; Rosenberg et al. 2013; Ma et al. 2003). Transfected HEK cells have already been used to produce recombinant coagulation factors which have been approved by FDA (Food and Drug Administration); however, full length mAbs produced by them are still awaiting approval (Lai et al. 2013; Berlec and Štrukelj 2013). Furthermore proprietary technologies, such as VelocImmune®, BiTE®, POTELLIGENT™, UltiMAb® and XenoMouse®, are used for production of monoclonal antibodies (Jakobovits et al. 2007; Murphy 2009; Nelson and Paulos 2015; Sheridan 2010; Shitara 2009). The mAb-derived products include fusion proteins, antigen binding fragments as well as composite proteins (Lefranc et al. 2009; Povey et al. 2001; Ecker et al. 2015; Li and Zhu 2010).

Generic monoclonal antibody-derived therapeutic structures as adapted from IMGT (Lefranc et al. 2009; World Health O 2006). Fc constant region which contributes to effector function, immune response and increased half-life, Fv variable region that contains complementarity determining regions (CDRs) facilitating antigen binding, Fab antigen binding fragment which lacks Fc region, scFv single chain fragment variable, FP Fc fusion proteins that contain Fc region for effector functionality (e.g. Abatacept), CP composite protein that contains Fc region for increasing half-life and not for effector functionality (e.g. Strensiq™) (World Health 2006)
MAbs: safety pharmacology and side effects
MAbs and related therapeutics are highly desirable from a biopharmaceutical perspective as they are highly target specific and well tolerated within the human system. Nevertheless, several mAbs have been discontinued or withdrawn based either on their inability to demonstrate efficacy and/or due to adverse effect, for example, Efalizumab, Biciromab and Fanolesomab, while others were discontinued due to high manufacturing costs, for example, Imciromab and Arcitumomab (Lefranc et al. 2009). Approved monoclonal antibodies as well as derived products have been associated with adverse effect, and these effects have been classified into categories of specialized toxicity as indicated in Table 1 (Peluso et al. 2013; Hansel et al. 2010). The reporting of these adverse effects is to be treated with caution as there are several factors that influence them, such as underlying conditions, drug combinations, reporting practices and clinical practice involved in the clinical trials.
The catastrophic TGN1412 clinical trial that resulted in multiple organ failure of six healthy volunteers reiterated the need for better preclinical safety testing. The underlying problems that were subsequently identified in this trial were mainly the lack of appropriate preclinical testing and model organisms chosen for study of adverse effects. The standard in vitro assays failed to capture the in vivo adverse effects in humans (Stebbings et al. 2013). In vivo toxicity studies using rodent or primate models are not always representative of the human system. Human therapeutics such as monoclonal antibodies are highly specific and targeted, and there is, therefore, a higher likelihood of false positive efficacy or false negative toxicity if such entities are tested in non-human models, both outcomes being highly undesirable.
Eloctate showed haemotoxicity and hepatotoxicity in animal studies (mice and monkeys), but none have been reported in human clinical trials (Lower 2015). TGN1412 did not show the pro-inflammatory cytokine storm in in vivo tests (cynomolgus macaques) due to the absence of CD28 on its CD4+ effector memory T cells as well as in in vitro tests (human lymphocytes) due to the lack of localization of cell receptor (Stebbings et al. 2013). There are different factors which can influence the safety and efficacy of mAbs. Binding affinity, glycoforms, valency and density of antigens as well as antibodies, cell surface receptor and binding interface are some of the factors that contribute to the biological activity of mAbs and, if suboptimal, could lead to reduction of efficacy or an increase in toxicity (Stebbings et al. 2013; Jefferis 2014). Nimotuzumab exhibits lower dermal toxicity due to optimal binding affinity to EFGR that ensures its binding below toxic levels (Boland and Bebb 2009).
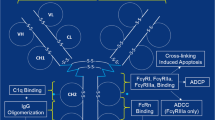
Effector functions of mAbs and related products, such as antibody-dependent cell phagocytosis (ADCP), antibody-dependent cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) as well as evoking other cell-mediated immune responses, are modulated via the Fc region by interaction with FcγR receptors on different immune responsive cells (Fig. 2a) (Carter 2006). This also regulates the pharmacokinetics, transcytosis, catabolism and placental transfer of antibodies via the FcRn (neonatal Fc Receptor) as summarized in Table 2 (Roopenian and Akilesh 2007). Glycosylation at the Fc region occurs at N297 and consists of a core heptasaccharide region comprising mostly N-acetylglucosamine and mannose residues as well as the variable region as seen in Fig. 2b (Carter 2006). Modifying the Fc region either via amino acid substitution or by a change in glycosylation pattern has shown to change effector functionality. IgG1-based therapeutic antibodies have shown increased ADCC and ADCP activity with substitution at amino acid positions 298,333 and 334, whereas Otelixizumab has shown reduced ADCP and ADCC activity with an N297A substitutions(Shields et al. 2001; Bolt et al. 1993).The mammalian cell production systems could alter the glycoform, and this could either change the effector function-mediated therapeutic activity or induce immunogenic effects of mAbs (Jefferis 2009). Afucosylation and bisecting N-acetylglucosamine were reported for antibodies produced in CHO cells, and they were associated with reduced ADCC activity (Shields et al. 2002; Umaña et al. 1999). Galactosylation levels are important for different functions, such as transport of IgG molecules across placenta and complement activation. Mammalian cell lines generally produce hypogalactosylated products; however, if this hypogalactosylation is unintended, it could impact effector function. This has been demonstrated with Alemtuzumab and rituximab where the removal of galactose residues reduced complement activation (Raju and Jordan 2012; Boyd et al. 1995). Mammalian production systems can also add oligosaccharides not present in human system, such as addition of N-glycolylneuraminic acid by CHO, NS0 and Sp2/0 systems, which can be immunogenic (Jefferis 2014).

a Monoclonal antibody structure with binding site for antigen, FcγR and FcRn receptor as well as glycosylation sites (Glycan); Ag antigen, CDC complement-dependent cytotoxicity, ADCC antibody-dependent cell cytotoxicity, ADCP antibody-dependent cell phagocytosis, b glycosylation profile at N297 residue of the Fc region of antibodies. The bold line indicates core structures, and dotted line indicates variable structures. Gal galactose, SA sialic acid, man mannose, GlcNAc N-acetylglucosamine, Fuc fucose, Asn asparagine (N297)
Although the trends seem to be in favour of biopharmaceutical development, the growth rates have not yet reached their full potential due to financial and technical complexities involved in early stages of research and development and preclinical testing as described in the following sections. The comprehensive costs of developing a new drug amount to $2.8 billion (Pharma 2014). Studies done over the past decade show that nearly 90 % of drugs failed in clinical development (66 % in Phase I and 30 % in Phase II) and this high attrition rate is the major contributing factor to the exorbitant cost of new drug development (Hay et al. 2014; Kola and Landis 2004; Paul et al. 2010). Thus, it is more beneficial to address attrition, as a 10–15 % decrease in attrition rate could reduce the cost of drug development by nearly 35 % (Paul et al. 2010). Recent studies reported that toxicity and lack of efficacy were the most important factors for high attrition rates in small molecule drug development (Waring et al. 2015). Unlike conventional drugs which mainly revolve around small molecule chemistry, biological drugs are far more complex to produce and characterize as they are 200–1000× larger, structurally more complex and highly sensitive to their manufacturing conditions. The costs involved in development and production of biopharmaceutical entities are 1.5–2.5× higher than that of small molecule drugs (Blackstone and Fuhr 2007). With nearly 80 % of biological drugs failing in clinical development mainly due to lack of efficacy and safety, there arises an urgent need for smarter preclinical development. This requires better product understanding, i.e. examining characteristics which contribute to product quality such as biological activity, affinity, pharmacology, toxicity, immunogenicity, thus leading to early prediction of success/failure. Improved product understanding and rapid screening of potential drug candidates by utilizing different in vitro and in silico methods to predict efficacy and safety techniques would lead to better preclinical design.
In vitro systems for toxicity testing
The general in vitro toxicity testing panel includes cellular, biochemical and molecular assays to study cytotoxicity, reactive oxygen species production as well as specialized toxicity effects including genotoxicity, hepatotoxicity, immunotoxicity to name a few. They are assessed via standard, specialized or target organ cell-based assays. Techniques such as WST, MTT, MTS, BrDu and Alamar blue are commonly used to asses basal cytotoxic or direct effect on cell proliferation, whereas Annexin V/Propidium iodide staining can help distinguish between necrotic and apoptotic events. Mitochondrial damage can be assessed by mitochondrial membrane potential assays and luminescent cell viability assays that quantify ATP. Protein marker-based techniques, such as assessing caspase cleavage via flow cytometry or western blotting techniques, can also be used to understand the mode of action of particular compounds. Reactive oxygen species production leads to oxidative stress, and this can also lead to cellular damage. There are different dyes, such as fluorescent and bioluminescent dyes, that can be utilized to study this effect. For gauging specialized toxicity effects, different types of biochemical, molecular and mode of action-based endpoints can be utilized. In vitro experimental data when combined with physicochemical properties and absorption, distribution, metabolism and elimination (ADME) characteristics help establish physiologically based pharmacokinetic (PBPK) and partitioning models (based on fundamental thermodynamic principles). Metabolism of parent compound, toxicity and likelihood of metabolites also allow for a more robust model to be developed as they help to take into account biotransformation and bioavailability. The above information helps to identify the doses and the class of compounds that have to be further tested in in vivo tests as specified by OECD guidelines for toxicity testing.
Monoclonal antibodies evoke an effector response mainly via antibody-dependent cytotoxicity, phagocytosis and complement-dependent cytotoxicity for eliminating tumour target cells (Kindt et al. 2007). For testing the biological activity of mAb-based therapeutics in vitro, the target cell line is cocultured with the molecule as well as effector cells derived either from PBMCs in human blood or cultured effector cells in a defined target to effector ratio (Golay et al. 2013). These effects can be studied by techniques which involve loading target cells with fluorescent membrane permeable dyes that are released upon target cell lysis. To assess mast cell degranulation, in vitro systems are incubated with drug of interest, and endpoints like histamine are then measured via spectroscopy or flow cytometry (Demo et al. 1999). Alternatively specific biomarkers like complement fragments can be used to detect specific events such as complement activation (Golay and Introna 2012). Cytokine release assays provide information about the extent and the kind of pro-inflammatory cytokine release. This is often assessed by introducing the monoclonal antibody to human lymphocytes and then assessing the supernatant for different types of cytokines, and this assay can often be performed in a multiplex format with flow cytometer analysis (Lash et al. 2006). A cytokine storm is a life-threatening adverse effect induced by monoclonal antibodies such as in the case of TGN1412 (Suntharalingam et al. 2006). Animal models utilized for assessing immunotoxicity involve lymph node proliferation assay, local lymph node assay and more recently the mouse drug allergy model though the predictive ability of these in vivo models have not been well characterized or validated (Whritenour et al. 2016). For assessing specialized toxicity assays, specific endpoints or biomarkers can be studied. Drug induced liver injury, liver enzyme inhibition or induction (particularly cytochromes 450, flavin monooxygenases and numerous others), change in human pregnane X Receptor activity as well as drug transporter activities for hepatotoxicity; Ames test for mutagenicity, in vitro single cell electrophoresis (comet) assay and DNA-based dyes for genotoxicity; human ether-related à-gogo gene related (hERG) assays, prolongation of QT interval, patch clamp assay, embryonic stem cell differentiation assay for cardiotoxicity and so on are examples used in small molecule drug development (Ekins 2014).
These issues regarding pharmacodynamics, selection of model organism, route of administration, dose, metabolism, toxicity studies have been addressed by the ICH Safety Pharmacology guideline S6 (R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. The safety pharmacology of mAbs, however, cannot be optimally assessed by standard toxicological assays alone (Cavagnaro 2002; Guideline 1997).
In silico tools for predictive toxicology
Computational toxicology tools could substantially aid in safety pharmacology testing of monoclonal antibody-derived therapeutics as they impart elements of automation, consistency and reliability to standard toxicological assays. There are a multitude of advantages offered by computational toxicology methods. They help to realize the 3R principle, i.e. replacement, reduction and refinement, by reducing the number of experimental animals used in drug safety testing. They also address the practical and economical concern of industries by providing a rapid and cost-effective way for safety testing of novel drug molecules. This in turn helps to cut down attrition rates and thus reduce the financial burden on the discovery and the development of new drugs. Furthermore, computational toxicology methods help to prioritize testing of those compounds which could be associated with toxic hazards by virtue of a problematic chemical space. This could be by means of structural similarity, indiscriminate interaction with closely related pharmacological targets and/or off target effect or other molecular events which are adaptable to in silico methods. Computational toxicology methods also prove useful when animal studies do not adequately represent the fate of drugs in humans (Ekins 2014; Cronin and Madden 2010; Greene and Pennie 2015; Wilson 2011).
Though these in vitro and in silico methods, such as physiologically based pharmacokinetic (PBPK) modelling and qualitative/quantitative structure–activity relationships (QSAR), are extensively used for predicting biological activity as well as toxicity during small molecule drug development (Table 3), their full potential has not been utilized for biological drug development.
Predictive model development
From the different in silico tools listed in Table 3, a summarized workflow for predictive toxicology model development is depicted in Fig. 3a. The main question to consider while developing a computational model is what can be modelled? The starting point of model development is data which can be of different types such as numeric, categorical, discrete or continuous and can be acquired from different sources like experiments, structures, physicochemical properties and so on. Algorithms are then required to preprocess these data as well as for feature extraction. This is mainly for selecting the inputs and outputs of models as well as to convert raw data into parameters that can be modelled mathematically, i.e. profilers or descriptors. Different linear and nonlinear mathematical techniques can be used for associating these descriptors to an adverse effect or toxicity by means of statistics, rules, multivariate data analysis and/or expert knowledge thus leading to development of a predictive model as shown in Fig. 3b. The resulting model must be validated to ensure non-discriminatory comparison with other existing models. Several factors would have to be taken into consideration while selecting a software platform/tool such as availability, accessibility, user expertise levels, transparency of algorithm and knowledge base, choice and complexity of methodology and inclusion of mechanistic elucidation. Performance would depend on choice of measures for robustness and goodness of fit as well as validation parameters and methods chosen. Some of these aspects are described in detail in the following sections keeping in mind the proteinaceous nature of mAbs.

a Computational toxicology model development workflow, b techniques involved in different types of predictive models
Databases
A number of databases have been utilized for developing predictive toxicology models during small molecule development such as Open TG GATEs, Pharmapendium, Drugmatrix® and ToxFX® (Greene and Pennie 2015). Databases containing information about mAbs and derived therapeutics are being developed extensively, and the IMGT mAb database is particularly noteworthy in this regard as it provides comprehensive information on structure, primary sequences, developmental status, targets as well as documents relating to approval for more than 589 entities (Lefranc et al. 2015). Sources like Drug Bank, patents, FDA documents and UniProt could yield useful information regarding sequences of mAbs, whereas Protein Data Bank (PDB) could provide structural information. The choice of a dataset for training model impacts its performance as studies have frequently indicated the discrepancies between public and proprietary datasets, i.e. performance of a model developed on public datasets is lower when applied on a proprietary dataset (Greene and Pennie 2015).
Descriptor generation and model development
Multivariate and statistical data analysis techniques have further allowed for rapid and easier descriptor calculation and model development. For proteins, the primary amino acid sequence and in some cases the 3D structure form the basis of generating different physicochemical, thermodynamic and topographic indices where the physicochemical and structural characteristics of amino acids are utilized to derive descriptors. These include principal component analysis-derived descriptors such as z scales and T-scales; 3D structure-based ones such as isotropic surface area and electronic charge index; atomic charge density-derived ones such as transferable atomic equivalent, to name a few (van Westen et al. 2013a, b). Several machine learning and statistical methodologies, such as support vector machines (SVM), artificial neural networks (ANNs), k-nearest neighbor approach (kNN), decision forest approach, Naïve Bayes, C4.5 decision tree, Bayesian models, random forest approaches, recursive partitioning, multiple linear regression (MLR), discriminant analysis (DA) and self-organizing maps (SOM), have been used to predict hepatotoxicity, genotoxicity, cardiotoxicity and renal toxicity of small molecules (Ekins 2014; Greene and Pennie 2015; Wilson 2011; Hardy et al. 2010). They can be used to build standalone inference-based models or combined with quantitative structure–activity relationship modelling.
Models
Quantitative structure–activity relationships (QSAR) approach is based on connecting an activity, in particular toxicity (QSTR) or any other property (QSPR), to descriptors which can be derived from physicochemical, structural, electronic or steric parameters (Hansch et al. 1995). QSAR methodology works best when the biological activity in question is based on a single endpoint or a simplistic mechanism of action. The development of QSAR models has been supported extensively by workflow tools, QSAR databases as well as uniform reporting and summarizing formats. Expert/Hybrid systems are extension of QSAR models, and they can be based on rules, knowledge or statistics as well as a combination of two or more approaches. The multivariate techniques used can either be linear, such as principal component analysis (PCA) or partial least square regression (PLS) used in TOPKAT, or nonlinear techniques, such as ANNs, used in CSgenoTox (Cronin and Madden 2010). Knowledge-based expert systems have incorporated a more mechanistic basis to their predictive tools (Cronin and Madden 2010). QSAM (quantitative sequence activity modelling) is another paradigm of QSAR modelling which is being used extensively for protein-based predictive models. Angiotensin-converting enzyme (ACE)-inhibitory peptides were screened based on models generated using PLS, MLR and most recently ANN (Zhou et al. 2008). PLS, SVM and HM-based models have been used with smaller peptides (9 amino acids residues) for predicting binding affinity with Class I Major Histocompatibility Complex (Zhao et al. 2007). Proteochemometric modelling is an extension of QSAR that uses multiplication of ligand and protein descriptors (MLPD) to include interaction space information in addition to protein and ligand descriptors (Qiu et al. 2016).
The advantages of QSAR-based expert systems are that they are rapid, well developed and regularly updated. The disadvantages are that the datasets, algorithms and knowledge base are usually not transparent. Most of the tools are commercial and use proprietary datasets. Due to the high level of automation, there is a possibility of losing the mechanistic understanding of action.
In addition to the models mentioned above, significant advances have been made with regard to ADME models as understanding the ADME characteristics of molecules is very important in assessing their bioavailability. A target mediate drug disposition-based pharmacokinetic model has been developed from preclinical data for predicting pharmacokinetics of mAbs within the human system which could aid in clinical designs (Luu et al. 2012). There have been several machine learning techniques that have been employed in skin absorption and metabolizing studies which enable to predict the extent of toxicity caused by compounds (Ashrafi et al. 2015; Moore et al. 2014). It is also worthwhile to mention that the latest techniques seem to revolve around consensus modelling where the outputs from different predictive models are averaged or inferred by several approaches, for example, leverage-weighted means (Cronin and Madden 2010). The success of these models, however, has been debatable as some report better predictivity, while others report no significant benefits when compared to single models (Hewitt et al. 2007).
Validation
Models are assessed for specificity, sensitivity and concordance based on either a different dataset typically referred to as the test set or by other appropriate means of validation. Internal validation procedures implemented include cross-validation (leave out one and/or leave out many) and bootstrapping. External and independent validation strategies can also be used such as testing the model with new experimental data. The predictive ability can be quantified using different parameters like root-mean-square error (RMSE), determination coefficient (R 2) and predictive squared correlation coefficient (Q 2) for QSAR model, and these have been evaluated in previous studies (Abshear et al. 2006; Consonni et al. 2009).
Discussion: status Quo and scope for mAb-based application
Different approaches have to be adopted for safety evaluation of monoclonal antibody-derived therapeutics when compared to small molecule drugs owing to innate differences like species specificity, degradation, increased half-life, complex dose–response relationship, interaction, lack of generic testing material, pleiotropic and synergistic mechanisms to name a few (Cavagnaro 2002).
Whether it is for assessing preclinical safety or for rapid screening, in vivo systems are not the most suitable models for studying the effects of monoclonal antibody-based therapeutics. The rationale behind using in vivo studies in preclinical safety testing is that the indirect immune-mediated response induced by the antibody as well as the magnitude of the effect cannot be gauged via standard in vitro tests. However, species specificity still remains the main obstacle. Studying the effector function becomes difficult due to differences in the FcγR receptors structure and affinity, complement system response and absence of target antigen (Golay and Introna 2012). Presence, number, interactions as well as distribution of target antigen also play an important role in assessing the biological activity of monoclonal antibodies (Golay et al. 2001). Attempts have been made to solve this problem by different strategies, such as knockout mice that lack mouse FcγR, transgenic mice expressing human FcγR, generating xenografts with human antigen in mouse cell lines, using completely mouse systems and using primate models such as rhesus monkey (Golay and Introna 2012; Barouch et al. 2013; Bournazos et al. 2014; Strasser et al. 2013). Animal testing is also expensive, sample size dependent and resource intensive. The main bottleneck in using in vitro systems for assessing the toxicity of mAbs is that the effector cells have to be coincubated or cocultured with the cell line of interest. The sensitivity and specificity of these assays depend on several factors which have to be optimized, such as cell density, incubation times as well as the choice of system and assay endpoint. The innate complexity, diversity and size of mAbs-based therapeutic as well as their diverse mechanisms of actions that involve many pathways exacerbate the need for carefully designed in vitro systems that take into account all of the above factors. In standard cytokine release assays, the mAbs bind to receptors all over the cell which is not an accurate representation of the human systems where cytokine release is sometimes dependent on localized receptor interaction (Stebbings et al. 2007). Sophisticated analytical techniques used in studying the endpoints of these assays have to be carefully assessed for resolution as well as sensitivity in detecting events as they can be prone to artefacts owing to nature of assay in question as well as the size of biological molecules. Artefacts can arise while using flow cytometry techniques due to homotypic adhesion as demonstrated with anti CD20 antibodies monoclonal antibodies (Golay et al. 2010). New generation preclinical safety testing tools would have to be high-throughput, rapid and cost-effective to meet the accelerated growth of the biopharmaceutical market. They also need to be highly reproducible and be fairly predictive to allow for rapid screening facilitating reliable selection of new compounds at initial stages thus saving time and money to allow more focus on drug development for rare diseases. They would also provide an alternative to animal testing considering the various drawbacks of in vivo systems as seen in the case of TGN1412. In vitro systems have now evolved from 2D cocultures to 3D spheroidal cocultures, organs on chips as well as whole blood systems to better mimic the responses that could be produced in a human system (Whritenour et al. 2016). Immunotoxicogenomics and expression profiling of both in vivo and in vitro systems are being used to identify pathways, mechanism of action as well as biomarkers for study of delayed hypersensitivity reactions (Shao et al. 2014). These advancements may contribute to better designed preclinical testing strategies for monoclonal antibody-derived therapeutics.
Appropriate and relevant experimental studies are of paramount importance in non-clinical safety testing as they also contribute to good datasets which can then be modelled. Most of the models are developed based on public datasets and fail to perform adequately when tested with proprietary datasets. The highly competitive nature of the biopharmaceutical industry makes information access very difficult. There are also difficulties in feature extraction for biological molecules owing to their complexity and size. The applicability of such modelling techniques in rapid screening depends on the experimental set-up as well as on identifying and forming sensible profilers and descriptors. Like all proteins, the primary sequence provides a wealth of information for mAbs. However, there is high degree of sequence similarity, especially in the Fc region, and this would mean that appropriate techniques such as benchmarking would have to be incorporated to select relevant descriptor sets (van Westen et al. 2013a, b). Descriptor for proteins molecules can be generated by different software such as PseAAC, Protein Recon, PROFEAT and ProtDCal, of which ProtDCal, a freely available tool with a friendly graphical user interface, has the capacity to generate a higher number of non-redundant of molecular descriptors for proteins from FASTA or PDB files (Ruiz-Blanco et al. 2015). Another possible concern is that primary sequence-based descriptors do not take into account neither interactions between amino acid residues nor the antibody-antigen and antibody-receptor interaction space. There are different modelling platforms for predicting antibody structures from primary sequences such as PIGS(Prediction of Immunoglobulin Structures), Rosetta antibody, Web Antibody Modelling (WAM) and Abysis databases among which PIGS performs better (Marcatili et al. 2014). RCSB integrates different bioinformatics and structural tools for comparison of primary and secondary structures. Advances made in PCM techniques include a new descriptor for antigen–antibody interaction called epitope–paratope interaction fingerprint (EPIF) which tries to address the higher time-complexity of MLPD, thus allowing for simplification the antigen–antibody interaction term (Qiu et al. 2015, 2016). Platforms like proABC, ABangle and LYRA allow for modelling antigen–antibody interactions, orientation of variable chain and lymphocyte receptor, respectively (Klausen et al. 2015; Olimpieri et al. 2013; Dunbar et al. 2013). Physicochemical characteristics of mAbs will influence PK/PD properties (increased binding to serum proteins and increased half-life) which affects ADME characteristics thus impacting bioavailability and biological activity. Glycosylation is another aspect that has to be taken into consideration as change in glycosylation pattern could affect functionality as well as impact PK/PD characteristics of mAbs (Liu 2015). Successful attempts have been made from a bioengineering point of view to investigate the effects of the production process on glycosylation profiles of monoclonal antibodies by using multivariate techniques, such as principal component analysis, partial least squares and parallel factor analysis (Green and Glassey 2015; Glassey 2012). Glycoengineered antibodies were produced by CHO cells with higher glycosyltransferase which enabled the production of engineered antibodies with the N-acetylglucosamine profiles required to achieve higher neutrophil-mediated phagocytosis activity and thus greater efficacy in killing tumour cells (Umaña et al. 1999; Golay et al. 2013). Indeed, engineered glycoforms of anti-CD20 antibodies, such as obinutuzumab and rituximab, have sevenfold higher binding affinity to neutrophils and thus an increased neutrophil-mediated phagocytosis-based killing of tumour cells (Golay et al. 2013). The challenge would then be to associate these attributes to potential adverse effects which will then allow for development of predictive toxicology models. Intricate algorithms would also be required for associating profilers and descriptors with synergistic endpoints of toxicity. Along with carefully designed experimental procedures, extensive expert knowledge would be required for such model development.
Conclusion
Biopharmaceuticals have positively impacted the lives of millions. They have paved the way for personalized medicines, improve prognosis of cancer, genetic and immune disorders as well as breakthroughs in rare disease management. The advances made are, however, impeded by a lack of progress in bioprocess development strategies as well as increasing costs owing to attrition, wherein the lack of efficacy and safety accounts for nearly 60 % of all factors contributing to attrition (Kola and Landis 2004). This reiterates the need for carefully designed predictive models to assess the efficacy as well as toxicity of potential drug candidates at an early stage. A more effective, high-throughput rapid screening of candidates based on adverse effects is required at an early stage to filter out the number of candidates proceeding to clinical trials. A choice of appropriate in vivo systems should be in place along with better proof of concept studies as animal models are not representative of human systems for assessing the efficacy and safety of biopharmaceuticals in specialized therapy areas like oncology and immunology. Alternative approaches such as specialized in vitro toxicology tests, better biomarkers and omics approaches can be utilized for this purpose. In this regard, computational toxicology tools like expert/hybrid systems provide a powerful complement to in vitro systems as they will allow for development of automated and reliable models for predicting toxicity or adverse effect of monoclonal antibody therapeutics. In order to make these predictive platforms more robust, descriptor calculation, feature extraction, inclusion of pharmacokinetics and bioavailability characteristics, mechanistic understanding and multidisciplinary expert knowledge will be of paramount importance. This will pave way for the development of rapid bioprocess development strategies for faster development of effective and safe biopharmaceuticals and may in fact change the face of biopharmaceutical manufacturing as we see today.
References
Abshear T, Banik GM, D’Souza ML, Nedwed K, Peng C (2006) A model validation and consensus building environment. SAR QSAR Environ Res 17:311–321
Ashrafi P, Moss GP, Wilkinson SC, Davey N, Sun Y (2015) The application of machine learning to the modelling of percutaneous absorption: an overview and guide. SAR QSAR Environ Res 26:181–204
Barouch DH, Whitney JB, Moldt B, Klein F, Oliveira TY, Liu J, Stephenson KE, Chang H-W, Shekhar K, Gupta S (2013) Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature 503:224–228
Berlec A, Štrukelj B (2013) Current state and recent advances in biopharmaceutical production in Escherichia coli, yeasts and mammalian cells. J Ind Microbiol Biotechnol 40:257–274
Boland WK, Bebb G (2009) Nimotuzumab: a novel anti-EGFR monoclonal antibody that retains anti-EGFR activity while minimizing skin toxicity. Exp Opin Biol Ther 9:1199–1206
Bolt S, Routledge E, Lloyd I, Chatenoud L, Pope H, Gorman SD, Clark M, Waldmann H (1993) The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol 23:403–411
Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV (2014) Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 158:1243–1253
Boyd PN, Lines AC, Patel AK (1995) The effect of the removal of sialic acid, galactose and total carbohydrate on the functional activity of Campath-1H. Mol Immunol 32:1311–1318
Carter PJ (2006) Potent antibody therapeutics by design. Nat Rev Immunol 6:343–357
Cavagnaro JA (2002) Preclinical safety evaluation of biotechnology-derived pharmaceuticals. Nat Rev Drug Discov 1:469–475
Consonni V, Ballabio D, Todeschini R (2009) Comments on the definition of the Q 2 parameter for QSAR validation. J Chem Inf Model 49:1669–1678
Cronin MTD, Madden JC (2010) In silico toxicology: principles and applications. Royal Society of Chemistry, London
Demo SD, Masuda E, Rossi AB, Throndset BT, Gerard AL, Chan EH, Armstrong RJ, Fox BP, Lorens JB, Payan DG (1999) Quantitative measurement of mast cell degranulation using a novel flow cytometric annexin-V binding assay. Cytometry 36:340–348
Dunbar J, Fuchs A, Shi J, Deane CM (2013) ABangle: characterising the VH–VL orientation in antibodies. Prot Eng Des Sel 26:611–620
Ecker DM, Jones SD, Levine HL (2015) The therapeutic monoclonal antibody marketed. MAbs 7(1):9–14
Ekins S (2014) Progress in computational toxicology. J Pharmacol Toxicol Methods 69:115–140
Glassey J (2012) Multivariate data analysis for advancing the interpretation of bioprocess measurement and monitoring data. In: Measurement, monitoring, modelling and control of bioprocesses. Springer, Heidelberg, pp 167–191
Golay J, Introna M (2012) Mechanism of action of therapeutic monoclonal antibodies: promises and pitfalls of in vitro and in vivo assays. Arch Biochem Biophys 526:146–153
Golay J, Lazzari M, Facchinetti V, Bernasconi S, Borleri G, Barbui T, Rambaldi A, Introna M (2001) CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood 98:3383–3389
Golay J, Bologna L, André P-A, Buchegger F, Mach JP, Boumsell L, Introna M (2010) Possible misinterpretation of the mode of action of therapeutic antibodies in vitro: homotypic adhesion and flow cytometry result in artefactual direct cell death. Blood 116:3372–3373
Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambaldi A, Klein C, Introna M (2013) Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 122:3482–3491
Green A, Glassey J (2015) Multivariate analysis of the effect of operating conditions on hybridoma cell metabolism and glycosylation of produced antibody. J Chem Technol Biotechnol 90:303–313
Greene N, Pennie W (2015) Computational toxicology, friend or foe? Toxicol Res 4:1159–1172
Guideline ICHHT (1997) Preclinical safety evaluation of biotechnology-derived pharmaceuticals. In: International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use
Hansch C, Hoekman D, Leo A, Zhang L, Li P (1995) The expanding role of quantitative structure-activity relationships (QSAR) in toxicology. Toxicol Lett 79:45–53
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJT (2010) The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov 9:325–338
Hardy B, Douglas N, Helma C, Rautenberg M, Jeliazkova N, Jeliazkov V, Nikolova I, Benigni R, Tcheremenskaia O, Kramer S (2010) Collaborative development of predictive toxicology applications. J Cheminform 2:1–29
Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J (2014) Clinical development success rates for investigational drugs. Nat Biotechnol 32:40–51
Hewitt M, Cronin MTD, Madden JC, Rowe PH, Johnson C, Obi A, Enoch SJ (2007) Consensus QSAR models: do the benefits outweigh the complexity? J Chem Inf Model 47:1460–1468
Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G (2007) From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol 25:1134–1143
Jefferis R (2009) Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov 8:226–234
Jefferis R (2014) Monoclonal antibodies: mechanisms of action. In: State-of-the-art and emerging technologies for therapeutic monoclonal antibody characterization volume 1 monoclonal antibody therapeutics: structure, function, and regulatory space, vol 1176. American Chemical Society, pp 35–68
Jones PT, Dear PH, Foote J, Neuberger MS, Winter G (1985) Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321:522–525
Kindt TJ, Goldsby RA, Osborne BA, Kuby J (2007) Kuby immunology. Macmillan, London
Klausen MS, Anderson MV, Jespersen MC, Nielsen M, Marcatili P (2015) LYRA, a webserver for lymphocyte receptor structural modeling. Nucleic Acids Res 43:W349–W355
Köhler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256:495–497
Kola I, Landis J (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3:711–716
Lai T, Yang Y, Ng SK (2013) Advances in mammalian cell line development technologies for recombinant protein production. Pharmaceuticals 6:579–603
Lash GE, Scaife PJ, Innes BA, Otun HA, Robson SC, Searle RF, Bulmer JN (2006) Comparison of three multiplex cytokine analysis systems: luminex, SearchLight™ and FAST Quant®. J Immunol Methods 309:205–208
Lefranc M-P, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane JM (2009) IMGT®, the international ImMunoGeneTics information system®. Nucleic Acids Res 37:L1006–L1012
Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T (2015) IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res 43:D413–D422
Li J, Zhu Z (2010) Research and development of next generation of antibody-based therapeutics. Acta Pharmacol Sin 31:1198–1207
Liu L (2015) Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci 104:1866–1884
Lower PATA (2015) Mechanism of action. Drugs
Luu KT, Bergqvist S, Chen E, Hu-Lowe D, Kraynov E (2012) A model-based approach to predicting the human pharmacokinetics of a monoclonal antibody exhibiting target-mediated drug disposition. J Pharmacol Exp Ther 341:702–708
Ma JKC, Drake PMW, Christou P (2003) The production of recombinant pharmaceutical proteins in plants. Nat Rev Genet 4:794–805
Marcatili P, Olimpieri PP, Chailyan A, Tramontano A (2014) Antibody modeling using the Prediction of ImmunoGlobulin Structure (PIGS) web server. Nat Protoc 9:2771–2783
Moore CA, Wilkinson SC, Blain PG, Dunn M, Aust GA, Williams FM (2014) Percutaneous absorption and distribution of organophosphates (chlorpyrifos and dichlorvos) following dermal exposure and decontamination scenarios using in vitro human skin model. Toxicol Lett 229:66–72
Murphy A (2009) VelocImmune: immunoglobulin variable region humanized mice. Recombinant antibodies for immunotherapy. Cambridge university, GB, pp 100–107
Nelson MH, Paulos CM (2015) Novel immunotherapies for hematologic malignancies. Immunol Rev 263:90–105
Olimpieri PP, Chailyan A, Tramontano A, Marcatili P (2013).Prediction of site-specific interactions in antibody-antigen complexes: the proABC method and server. Bioinformatics btt369
Peluso R, Cafaro G, Di Minno A, Iervolino S, Ambrosino P, Lupoli G, Di Minno MND (2013) Side effects of TNF-α blockers in patients with psoriatic arthritis: evidences from literature studies. Clin Rheumatol 32:743–753
Pharma E (2014) World preview 2014, outlook to 2020
Povey S, Lovering R, Bruford E, Wright M, Lush M, Wain H (2001) The HUGO gene nomenclature committee (HGNC). Hum Genet 109:678–680
Qiu T, Xiao H, Zhang Q, Qiu J, Yang Y, Wu D, Cao Z, Zhu R (2015) Proteochemometric modeling of the antigen-antibody interaction: new fingerprints for antigen, antibody and epitope–paratope interaction. PLoS One 10:e0122416
Qiu T, Qiu J, Feng J, Wu D, Yang Y, Tang K, Cao Z, Zhu R (2016) The recent progress in proteochemometric modelling: focusing on target descriptors, cross-term descriptors and application scope. In: Briefings in bioinformatics, bbw004
Raju TS, Jordan RE (2012) Galactosylation variations in marketed therapeutic antibodies. MAbs 4(3):385–391
Roopenian DC, Akilesh S (2007) FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 7:715–725
Rosenberg Y, Sack M, Montefiori D, Forthal D, Mao L, Hernandez-Abanto S, Urban L, Landucci G, Fischer R, Jiang X (2013) Rapid high-level production of functional HIV broadly neutralizing monoclonal antibodies in transient plant expression systems. PLoS One 8:e58724
Ruiz-Blanco YB, Paz W, Green J, Marrero-Ponce Y (2015) ProtDCal: a program to compute general-purpose-numerical descriptors for sequences and 3D-structures of proteins. BMC Bioinform 16:1
Shao J, Berger LF, Hendriksen PJM, Peijnenburg AACM, van Loveren H, Volger OL (2014) Transcriptome-based functional classifiers for direct immunotoxicity. Arch Toxicol 88:673–689
Sheridan C (2010) Fresh from the biologic pipeline [mdash] 2009. Nat Biotechnol 28:307–310
Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B (2001) High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J Biol Chem 276:6591–6604
Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SHA, Presta LG (2002) Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem 277:26733–26740
Shitara K (2009) [Potelligent antibodies as next generation therapeutic antibodies]. Yakugaku zasshi. J Pharm Soc Jpn 129:3–9
Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I (2007) “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol 179:3325–3331
Stebbings R, Eastwood D, Poole S, Thorpe R (2013) After TGN1412: recent developments in cytokine release assays. J Immunotoxicol 10:75–82
Strasser A, Harris AW, Vaux DL, Webb E, Bath ML, Adams JM, Cory S (2013) Abnormalities of the immune system induced by dysregulated bcl-2 expression in transgenic mice. Curr Top Microbiol Immunol 166:175–181 (1990b)
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N (2006) Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355:1018–1028
Umaña P, Jean-Mairet J, Moudry R, Amstutz H, Bailey JE (1999) Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat Biotechnol 17:176–180
van Westen GJP, Swier RF, Wegner JK, Ijzerman AP, van Vlijmen HWT, Bender A (2013a) Benchmarking of protein descriptor sets in proteochemometric modeling (part 1): comparative study of 13 amino acid descriptor sets. J Cheminform 5:1
van Westen GJP, Swier RF, Cortes-Ciriano I, Wegner JK, Overington JP, Ijzerman AP, van Vlijmen HWT, Bender A (2013b) Benchmarking of protein descriptor sets in proteochemometric modeling (part 2): modeling performance of 13 amino acid descriptor sets. J Cheminform 5:1
Varma A, Cuenca J, Zhu Y (2014) Compostions and methods for producing glycoproteins. Google patents
Whritenour J, Casinghino S, Collinge M, Zhu X (2016) Nonclinical tools to assess risk of drug hypersensitivity reactions. Annu Rev Pharmacol Toxicol 56:561–576
Wilson AGE (2011) New Horizons in Predictive Toxicology: Current Status and Application. Royal Society of Chemistry, London
World Health O (2006) International nonproprietary names (INN) for biological and biotechnological substances. INN Working document 5:1–29
Zhao C, Zhang H, Luan F, Zhang R, Liu M, Hu Z, Fan B (2007) QSAR method for prediction of protein-peptide binding affinity: application to MHC class I molecule HLA-A* 0201. J Mol Graph Model 26:246–254
Zhou P, Tian F, Wu Y, Li Z, Shang Z (2008) Quantitative sequence-activity model (QSAM): applying QSAR strategy to model and predict bioactivity and function of peptides, proteins and nucleic acids. Curr Comput Aided Drug Des 4:311–321
Zhu J (2012) Mammalian cell protein expression for biopharmaceutical production. Biotechnol Adv 30:1158–1170
Acknowledgments
The authors gratefully acknowledge the financial support from the EU-Horizon 2020 Marie Skłodowska-Curie actions (MSCA) ITN project BIORAPID (No. 643056) and declare no competing interests. The authors’ affiliations are as shown on the cover page. The authors have full responsibility for writing and content of the paper. The authors gratefully acknowledge the financial support from the EU-Horizon 2020 Marie Skłodowska-Curie actions (MSCA) ITN project BIORAPID (No. 643056) and declare no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kizhedath, A., Wilkinson, S. & Glassey, J. Applicability of predictive toxicology methods for monoclonal antibody therapeutics: status Quo and scope. Arch Toxicol 91, 1595–1612 (2017). https://doi.org/10.1007/s00204-016-1876-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-016-1876-7