
Abstract
Non-hemolytic enterotoxin (NHE), a tri-partite, proteinaceous toxin encoded by contiguous nheA, nheB and nheC genes of Bacillus cereus sensu lato (B. cereus s.l.), is considered to be associated with the foodborne diarrheic syndrome. However, B. cereus s.l. includes a number of closely related strains, and the occurrence of NHE among them, and other members of Bacillus is unclear. Consequently, we aimed to determine the distribution and evolution of NHE within Bacillus by confirming the presence of the nheA, B and C sequences and variation within them using published data, and to analyze the genomic and genetic diversity. The phylogenetic tree of NHE proteins (NheA, NheB and NheC) from 81 different B. cereus s.l. strains was constructed. And on the genetic determinants of the NHE toxin did not bring any obvious link between the nheABC genes sequence of a strain and its virulence in the diarrhoeal pathogenesis. Analysis of the genomic diversity of the nheA, B and C loci revealed that their upstream regions were more conserved than the downstream sequences. Multilocus sequence typing schemes (MLST) based on seven concatenated housekeeping genes and nheA, B and C genes of the 75 strains were developed. The neighbor joining phylogenetic tree based on seven housekeeping genes together with nheA, B and C genes was similiar to published Bacillus phylogenetic trees. And on the genetic determinants of the NHE toxin did not bring any obvious link between the nheABC genes sequence of a strain and its virulence in the diarrhoeal pathogenesis.The results indicate that nheA, B and C genes do not affect the diversity of housekeeping genes, and this specific NHE protein does not participate in the diarrheic syndrome.
Similar content being viewed by others

Introduction
Bacillus cereus s. l. is widely distributed in food, soil and plants (Okinaka and Keim 2016). This Gram-positive, spore-forming bacterium may behave as an opportunistic human pathogen. Long known to be responsible for two forms of food poisoning, characterized by either diarrhea or nausea and vomiting. The diarrhoeal symptom includes the following symptoms, which usually last generally less than 48 h: abdominal pain, profuse watery diarrhea, sometime nausea and vomiting within 8–16 h. Although most cases are generally mild, more serious and even lethal cases have been reported in Europe.
Concerning the diarrhoeal syndrome, no definitive hypothesis that correlates the symptoms to a unique component exists to date. Indeed, several putative enterotoxins have been reported to be potentially responsible, alone or in combination, of the diarrhoeic pathotypes. These include the tripartite enterotoxins as Haemolysin BL (HBL) and Nonhaemolytic enterotoxin (NHE), but also the single-component toxin, Cytotoxin K (CytK) also sometimes named Haemolysin IV (HlyIV). In addition to these three major candidates, other molecules are also cited as potential enterotoxins involved in the diarrhoeic syndrome, such as enterotoxin FM (EntFM) (Boonchai et al. 2008), enterotoxin S (entS), enterotoxin-T (bceT) and pore-forming haemolysins like the Cereolysin O (CerO), Haemolysin II (HemII) and Haemolysin III (HlyIII). Besides, other virulence factors seem to contribute to the B. cereus foodborne diseases, such as phospoholipases or the sphingomyelinase (SMase).
The non-hemolytic enterotoxin NHE is encoded by nheA, nheB and nheC (Kim et al. 2015). NHE is a tripartite pore-forming toxin that requires the combination of three proteins: NheA, NheB and NheC. NHE was first isolated from the supernatant of a B. cereus s.l. culture that caused a large food poisoning outbreak in Norway in 1995 (Lund and Granum 1996). NHE proteins are secreted independently and maximal toxic activity on Vero cells requires all the three parts in a molar ratio 10:10:1 of NheA, NheB and NheC, respectively. NheB is the binding component of the enterotoxin complex and an increase in the concentration of NheC results in a decrease in Nhe toxic activity (Lindbäck et al. 2004).
However, the genes coding for these putative enterotoxins are, for the most part, largely distributed among the B. cereus group isolates, irrespective of their diarrheic activities (McIntyre et al. 2008; Swiecicka et al. 2006). Moreover, with the exception of the rabbit ileum assay, no animal model can be used to specifically test for the diarrhoeal properties of the strains (or for purified proteins) (Beecher et al. 1995). Only classical assays on animal cell lines are readily available, but they only give information on the generic cytotoxicity of these putative enterotoxins (Jeßberger et al. 2014).
B. cereus s.l. includes a number of closely related species, viz. Bacillus cereus sensu stricto, Bacillus mycoides, Bacillus pseudomycoides, Bacillus thuringiensis, Bacillus weihenstephanensis, Bacillus anthracis and Bacillus cytotoxicus, and many strains thereof (Lindbäck et al. 2004). The occurrence of NHE within the group, and other members of Bacillus is unclear, e.g., we have found NHE in some non- B. cereus s.l. members. Analysis methods such as MLST indexes the sequence variation present in a small number (usually seven) of housekeeping gene fragments located around the bacterial genome, is to provide a highly discriminating typing system that can be particularly helpful for the typing of bacterial pathogens (Keith and Martin 2014). And PHYLOViZ software make the data easy to be visualized and export the results in graphic formats (Alexandre et al. 2012). In this study, we were interested in determining the particular strains that produce NHE and to trace the molecular evolution and variation of the nheABC genes and the phylogenetic relationship of the various Bacillus cereus s.l. strains to others within Bacillus. Our approach involved sequence-based typing analysis, interrogation of online databases of allelic profiles and associated epidemiological data collected.
With the aim of assessing the potential implication of NHE in the diarrheic syndrome, the genomic and genetic diversity, as well as the occurrence and the evolutionary ecology of nheABC were studied in detail on a collection of Bacillus cereus strains.
Materials and methods
Bioinformatics and strains information
The presence of nheABC genes and their corresponding putative sequences was screened in the NCBI database among the 174 genomes from the B. cereus group as available (Aug. 15, 2015). The NheA, B and C protein sequences from B. cereus ATCC14579 were applied to find all the homologous proteins among the 174 strains by BLAST program in http://blast.ncbi.nlm.nih.gov/Blast.cgi.
A set of strains (Table 1) coming from food poisonings (FP), food products (F), the environment (E), clinical cases (C) and unknown origin (U) was collected to investigate the presence of nheABC genes. Then, a panel of positive strains was selected for MLST analysis (Table 2 and below). This selection was based on the origin, the year and the country of strains and the species to obtain the most diversified panel of nheABC positive strains.
MLST analysis
Seven loci encoding housekeeping genes were chosen for MLST analysis: glp (glycerol kinase), gmk (guanylate kinase), ilv (isoleucine-valine), pta (phosphate acetyltransferase), pur (purine synthesis), pyc (pyruvate carboxylase), tpi (triosephosphate isomerase) (Lampe and English 2016). 75 strains for which the nheABC genes are available in the genome databases, were selected for the MLST analysis (Table 2). The MLST result were shown in minimum-spanning tree (http://pubmlst.org/analysysis/). The nucleotide sequence diversity of glp, gmk, ilv, pta, pur, pyc, tpi and nheABC genes was analyzed at two levels: by constructing a phylogenetic tree for each loci with the CLC Main Workbench 7 software (ClCbio, a Qiagen Company) using the neighbor joining (NJ) algorithm with Jukes Cantor as substitution rate model, and by building sequence types (ST) of the various strains using the non-redundant database (NRDB) for allele comparison (http://pubmlst.org/). To cluster the strains according to their ST, BURST analysis was performed which defines a group when at least 5/7 loci were identical (http://pubmlst.org/perl/mlstanalyse/). Sequences of the seven chromosomal loci were also concatenated with or without adding the nheABC genes to construct and compare the respective phylogeny trees generated by CLC Main Workbench 7 software using UPGMA algorithm and Kimura 80 mathematical model using. The correctness of the results was evaluated using a 100-step bootstrap test (Virginie et al. 2015).
Results and discussion
nheABC occurrence among the B. cereus s.l. strains
While the mechanisms involved in the B. cereus diarrhoeal pathogenesis are still largely unknown, a large panel of enterotoxins have been designated as potential causative agents led to this syndrome. And NHE is regularly cited as a candidate because of its cytotoxic, necrotic and haemolytic activities on human intestinal cell lines (Lindback et al. 2004; Zhu et al. 2016). Some B. cereus genomes harbor the nhe operon which codes for the cytolytic protein NheA and the binding components NheB and NheC (Wehrle et al. 2009). In this study, the nheABC sequence from B. cereus ATCC14579 were applied to find all the homologous genes among the 174 strains by BLAST program in http://blast.ncbi.nlm.nih.gov/Blast.cgi. Since the first bacterial genome sequence was completed in 1995, 174 sequences of Bacillus strains genomes have been published in NCBI database until Aug. 2015 (the number of assembled and annotated B. cereus genome is 294 until Dec. 2016). The nheABC loci were found in 81 strains, including all the 31 B. anthracis strains, 24 B. cereus strains, 21 B. thuringiensis strains, 2 B. weihenstephanensis strains, 1 Bacillus bombysepticus strain and 1 Bacillus toyonensis strain. 2 B. mycoides strains were sequenced but only one strain contains nheABC loci.
However, Bacillus amyloliquefaciens, Bacillus atrophaeus, Bacillus clausii, Bacillus coagulans, Bacillus infantis, Bacillus lehensis, Bacillus licheniformis, Bacillus megaterium, Bacillus methanolicus, Bacillus methylotrophicus, Bacillus pseudofirmus, Bacillus pumilus, Bacillus sp., Bacillus subtilis, Bacillus halodurans, Geobacillus kaustophilus and Bacillus cellulosilyticus do not contain nheABC loci (Table 1). The nheABC operon occurrence in NCBI database is 46.6% (81/174). And the mean value frequency of nheA, nheB, nheC found in the literature is 82, 81 and 78%, respectively (Swiecicka et al. 2006; Gaviria et al. 2000; Hansen and Hendriksen 2001; Banerjee et al. 2011; Moravek et al. 2004; De Jonghe et al. 2010; Krause et al. 2010; Zhou et al. 2010; Samapundo et al. 2011; Chon et al. 2015). These frequencies are slightly higher than the ratio we measured from database. Maybe because more and more Bacillus genomes sequencing are completed, and nheABC operon is absent from the recently released genomes, so the ratio measured from database is low.
Genomic diversity of nheABC
81 genetic regions (30 kb in size) from the nheABC + strains mentioned above centered on the nheABC operon were collected and aligned for genomic diversity analysis. The putative ORFs (more than 200aa) from all fragments were annotated (Table 3), and compared to illustrate their genetic features. By using the nheABC locus of B. cereus ATCC14579 as a typical reference, good conservation was observed in the upstream region of this gene cluster among the other 76 strains. Interestingly, the rest four strains showed different pattern of conservation, therefore, we studied the genomic diversity in two parts.

In the first part, 30 kb DNA sequences centered on the nheABC loci from 76 strains were aligned and labelled in the sketch map. The alignment result showed a higher degree of conservation in the upstream region in terms of gene content, relative to the downstream region (Fig. 1). The downstream of nheABC locus contained six distinct conserved regions, which was different from their gene contents and organizations (indicated as A to F, and their relative frequencies of 5/76, 24/76, 2/76, 12/76, 31/76 and 2/76, respectively). Specifically, Branch A contained B. thuringiensis YBT-1520, HD-29, HD-1, HD-73 and B. mycoides ATCC6462. Branch B contained B. cereus AH820, 03BB102, AH187, ATCC10987, etc. strains. Branch C contained B. cereus NC7401 and Q1. Branch D contained B. cereus ATCC14579, AH187, 03BB102, etc. strains. Branch E contained all the 31 B. anthracis strains. Branch F contained B. weihenstephanensis KBAB4 and WSBC10204.

Genomic diversity of the 30 kb fragments centered on nheABC locus for forty-five B. cereus s.l. group strains (including 10 Ba, 22 Bc, 11 Bt, 1 Bm and 1 Bw). The alignment of the available sequences in database revealed a much higher degree of conservation in the upstream region in terms of gene content, compared to the downstream region. Orthologous genes are indicated by arrows of the same size and matches are indicated in gray shadow
In the nheABC locus upstream regions (left part of nheABC loci in Fig. 1), seven genes (two-component response regulator vanR, C terminus of sensor histidine kinase, M24/M37 family peptidase, manganase transport protein MntH, sulfur transferase, hypothetical protein and 2-dehydropantoate 2-reductase) were present in all the 76 fragments. Interestingly, in 49 of the 76 strains, a 505aa ORF (amino acid permease) was found immediately upstream of the NheA, while it was broken up into two smaller ORFs (also annotated as amino acid permease) in the 32 remaining strains, including which included 31 B. anthracis strains plus B. thuringiensis 97-27 strain.
In the nheABC locus downstream regions (right part of nheABC loci in Fig. 1), there was obvious genes constituent change and rearrangement in different branch. The gene rhtB (homoserine/threonine efflux protein) was conserved in Branch A–D, but disappeared in Branch E and F. The four genes cluster deoR (deoxyribonucleoside regulator), deoC (deoxyribose-phosphate aldolase), nupC (nucleoside transporter), pdp (pyrimidine-nucleoside phosphorylase) was well kept in the same direction in Branch B–F. And the gene yndJ encoded a putative membrane protein also maintained in Branch D and E. The other genes showed different variety. In the downstream regions, the composition and arrangement of genes diversified in different Branch strains.
But the rest five nheABC + strains, B. cereus ATCC4342, D17, FT9, G9241 and B. cytotoxicus NVH391-98, sequences showed high genomic diversity, could not be included into any branch.
To illustrate genes distribution of the whole 30 kb fragments in different branch, six representative strains were selected. Based on the 5.7 kb fragment from B. cereus NVH883-00, genes arrangement surrounding the nheABC locus from B. anthracis Ames with the counterparts from B. mycoides Rock1-4, B. pseudomycoides DSM12442, B. mycoides Rock 3-17, B. cereus NVH391-98 were shown in Fig. 2. All orthologous genes are indicated by arrows of the same size and matches are indicated in gray in the sketch map. B. mycoides Rock1-4, B. pseudomycoides DSM12442 and B. mycoides Rock 3-17 had almost the same genes distribution. It is easy to recognize gene cluster rearrangement and gene insertion in B. anthracis Ames and B. cereus NVH391-98, though these two strains shared high similarity in most genes rearrangement.

Comparison of the 30 kb fragment surrounding the nheABC locus from B. anthracis Ames with the corresponding nheABC regions from B. mycoides Rock1-4, 3-17, B. pseudomycoides DSM12442, B. cereus NVH391-98 and a 5.7 kb fragment from B. cereus NVH883-00. Orthologous genes are indicated by arrows of the same size and matches are indicated in gray
Genetic diversity of NheA, B and C
To further explore the genetic diversity based on the 81 collected NheA, B and C protein sequences, each protein was analyzed, respectively. The size of 73 NheA proteins was 386aa, and the size of the rest eight strains, such as B. cytotoxicus NVH391-98, B. cereus G9241, FT9, Ba HYU01, SVA11, RA3, B. anthracis BA1035, 2002013094 was 387aa, 312aa, 279aa, 388aa, 388aa, 388aa, 388aa and 318aa, respectively. Based on sequences diversity, all the 73 NheA proteins were divided into four groups (Fig. S1). In group A, the NheA from different strains shared 97–100% identities with B. cereus ATCC14579-NheA. In group B, the NheA from strains B. cereus G9241, Bw WSBC10204, KBAB4, Ba 2002013094, B. toyonensis BCT-7112 and Bm ATCC6462 shared 97, 96, 97, 97, 96 and 95% identities with B. cereus ATCC14579-NheA, respectively. In group C, the 312aa NheA from strain B. cereus G9241 shared 97% identity with B. cereus ATCC14579-NheA. Moreover, the coverage of B. cereus G9241 and Ba 2002013094 was only 78 or 82%, respectively. In group D, the 387aa NheA proteins from B. cereus NVH391-98 shared 78% identity with B. cereus ATCC14579-NheA.
NheB genetic diversity seemed more conservative. The size of 79 NheB proteins was 402aa, and the size of the rest two strains (B. cytotoxicus NVH391-98, B. cereus CI) was 401aa. Based on sequences diversity, all the 81 NheB sequences were divided into three groups (Fig. S2). In group A, the NheB from different strains shared 99–100% identities with B. cereus ATCC14579-NheB. In group B, the NheB from strains B. mycoides ATCC6462 shared 98% identity with B. cereus ATCC14579-NheB. In group C, the 401aa NheB proteins from B. cereus NVH391-98 shared 87% identity with B. cereus ATCC14579-NheB.
NheC genetic diversity analysis showed much less conservation, compared to NheA and NheB. The NheC size from the 81 strains ranged from 305aa to 397aa. Most of the proteins (74 out of 81) were 359aa. The size of the rest seven strains, such as Ba Vollum, CDC684, Han, B. cytotoxicus NVH391-98, B. cereus 4342, FT9 and B. thuringiensis AI Hakam was 305aa, 305aa, 353aa, 353aa, 362aa, 362aa, 397aa and 362aa, respectively. Based on sequences diversity, all the 81 NheC were divided into three groups (Fig. S3). In group A, the NheC from different strains shared 94–100% identities with B. cereus ATCC14579-NheC. In group B, the NheC proteins from Bw WSBC10204 and KBAB4 both shared 92% identity with B. cereus ATCC14579-NheC. In group C, the NheC from Ba Han shared 86% identity with B. cereus ATCC14579-NheC. In group D, the NheC protein from B. cereus NVH391-98 shared 73% identity with B. cereus ATCC14579-NheC.
In all the analysis of NheA, B and C genetic diversity, the strain B. cereus NVH391-98 was always exclusively different from other strains. This is in agreement with the observations performed on the genomic diversity of nheABC loci (Böhm et al. 2015).
Seven housekeeping genes MLST analysis from 75 nheABC + strains
To further characterize the sequence variation among the 75 nheABC positive strains of B. cereus, MLST was performed using the seven housekeeping genes: glp, gmk, ilv, pta, pur, pyc and tpi. The sequence variability of each locus was also studied in details. Based on the allelic profiles of the seven loci, ST could be defined for all the isolates. The high number of ST is another illustration of the high diversity existing among the nheABC positive B. cereus s.l. strains. Of note, Table S1 displays the correspondence between the allelic profiles defined in this work and those reported in previous MLST schemes (Helgason et al. 2004; Sorokin et al. 2006). The 75 isolates (Table S1) were then subjected to a BURST analysis to group the strains according to the similarity of their allelic profile. The isolates were grouped together when five out of the seven analyzed loci were identical. Based on this criterion, seven clusters were formed, as shown in Table 2. 75 strains were clustered in seven groups. Strains coming from foodborne outbreaks spread in different groups. This result showed that there was no obvious association between similar allelic profiles and geographic or source origins.
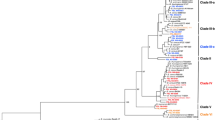
To visualize the influence of nheABC gene on the relatedness of the strains, 75 strains displaying nheABC positive (originating from different countries) were selected for the MLST analysis of the Bacillus strains. A tree was built with the sequences of the seven loci concatenated with nheABC. PHYLOViZ was built upon the goeBURST implementation (available at http://goeburst.phyloviz.net) and it allows to integrate and display multiple sources of information. The result was showed in Fig. 3. The whole tree was built on all the selected species about 2033 Bacillus cereus strains from different sources (data not shown), and the 75 selected strains assigned black color were located in this split tree. Though the observed frequency of each strain was different, all the selected strains spread randomly in all the part of MLST split tree. The number of locus differences between each pair strains could be observed from the line distances of the tree. This explained that the nheABC sequences variation had no favor of any type of strains, and also showed again that there was no obvious association between similar allelic profiles and geographic or source origins.

MLST split tree of Bacillus cereus. The location of each strain illustrated the number of locus differences with other strains in MLST split tree. The distances represented the number of locus differences between every pair of samples. The size of the circles indicated the observed frequencies. The black circles represented the strains from Table S1, the blue and green circles were 2033 Bacillus cereus strains (data not shown). The numbers in the circles were ST of each strains. a the whole tree. b the partial enlargement of A
To verify that nheABC operon was not prone to lateral transfer, 30 kb genome fragments centered on the nheABC operon locus were investigated. Although the genomic neighborhood of nheABC showed some variability, no element potentially involved in horizontal transfer was found (data not shown). The comparison of the 76 Bacillus fragments has provided valuable insights into defining the key genetic complement of the organisms, which forms the basic genetic support to define the organism’s pathogenicity ability (Lisdawati et al. 2015).
To investigate the genetic diversity of nheABC and seven housekeeping genes (glp, gmk, ilv, pta, pur, pyc and tpi), the concatenated sequences of the seven housekeeping genes (I), nheA, B and C genes (II), the concatenated sequences of the seven housekeeping genes plus the nheA, B and C genes (III) were collected from 35 B. cereus s.l. group strains for which the seven genes sequences were all available in NCBI database. The sequences were then analyzed by online software clustalw (http://www.genome.jp/tools/clustalw/). The neighbor joining trees based on seven housekeeping genes together with nheA, B and C genes were built. In tree I (Fig. 4), B. cereus NVH391-98 was far away from all the other 35 strains. Genes from different strains clustered together, according to the genetic relationship five main clusters are noted as A–E. In tree II and III, though the order of five main clusters changed, the constituent of each cluster stayed the same. The structure of these trees were similiar to published Bacillus phylogenetic trees (Virginie et al. 2015). This result indicated that no striking differences was observed between the various trees, and meant that the influence of nheABC on strain clustering is limited. This result also showed that the genetic determinants of the NHE had no any obvious relationship with the nheABC genes sequence of a strain and its virulence in the diarrhoeal pathogenesis.

The NJ trees built based on the concatenated sequences of the six housekeeping genes and nheABC genes. I (based on six housekeeping genes), II (based on nheABC genes) and III (based on the concatenation of six housekeeping genes and nheABC genes)
In this study, we found that the genetic determinants of the NHE toxin did not bring any obvious link between the nheABC genes sequence of a strain and its virulence in the diarrhoeal pathogenesis. To assess whether NHE is a significant factor in this disease, a transcriptomic study should be considered to take the genes expression of the toxin into account. And the potential action of NHE also should be investigated in concert with other possible enterotoxins (e.g., HBL, CytK or HlyII) and other virulence factors to evaluate the diarrhoeic potential of B. cereus strains. To elucidate the actual involvement of these molecules in the diarrhoeal syndrome, it is necessary to find an adequate animal model. In fact, due to their proteinaceous nature, these putative enterotoxins may be prone to a rapid inactivation in the intestinal tract, unless they would be released by the B. cereus cells in the immediate vicinity of host’s intestinal epithelium being protected by the mucus layer. However, this hypothesis has still to be verified.
Conclusions
The nheABC genes do not affect the diversity displayed by housekeeping genes, and this specific protein is probably not implicated in the diarrheal syndrome. Our data provide a scientific basis for us to know more about nheABC loci in food poisoning B. cereus.
References
Alexandre P, Francisco, Catia V et al (2012) PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics, 13:87–92.
Banerjee M, Nair GB et al (2011) Phenotypic & genetic characterization of Bacillus cereus isolated from the acute diarrhoeal patients. Indian J Med Res 133:88–95
Beecher DJ, Schoeni JL, Wong AC (1995) Enterotoxic activity of hemolysin BL from Bacillus cereus. Infect Immun 63(11):4423–4428
Böhm ME, Huptas C, Krey VM et al (2015) Massive horizontal gene transfer, strictly vertical inheritance and ancient duplications differentially shape the evolution of Bacillus cereus enterotoxin operons hbl, cytK and nhe. BMC Evol Biol 15:246
Boonchai N, Asano SI, Bando H et al (2008) Study on cytotoxicity and nucleotide sequences of enterotoxin FM of Bacillus cereus isolated from various food sources. J Med Assoc Thai 91(9):1425–1432
Chon JW, Yim JH, Kim HS et al (2015) Quantitative prevalence and toxin gene profile of Bacillus cereus from ready-to-eat vegetables in South Korea. Foodborne Pathog Dis 12(9):795–799
De Jonghe V, Coorevits A, De Block J et al (2010) Toxinogenic and spoilage potential of aerobic spore-formers isolated from raw milk. Int J Food Microbiol 136(3):318–325
Gaviria Rivera AM, Granum PE et al (2000) Common occurrence of enterotoxin genes and enterotoxicity in Bacillus thuringiensis. FEMS Microbiol Lett 190(1):151–155
Hansen BM, Hendriksen NB (2001) Detection of enterotoxic Bacillus cereus and Bacillus thuringiensis strains by PCR analysis. Appl Environ Microbiol 67(1):185–189
Helgason E, Tourasse NJ, Meisal R et al (2004) Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Appl Environ Microbiol 70:191–201
Jeßberger N, Dietrich R, Bock S (2014) Bacillus cereus enterotoxins act as major virulence factors and exhibit distinct cytotoxicity to different human cell lines. Toxicon 77:49–57
Keith A Jolley, Martin CJ Maiden (2014) Using multilocus sequence typing to study bacterial variation: prospects in the genomic era. Future Microbiol 9(5):623–630
Kim MJ, Han JK, Park JS et al (2015) Various enterotoxin and other virulence factor genes widespread among Bacillus cereus and Bacillus thuringiensis strains. J Microbiol Biotechnol 25(6):872–879
Krause N, Moravek M et al (2010) Performance characteristics of the Duopath(R) cereus enterotoxins assay for rapid detection of enterotoxinogenic Bacillus cereus strains. Int J Food Microbiol 144(2):322–326
Lampe BJ, English JC (2016) Toxicological assessment of nattokinase derived from Bacillus subtilis var. natto. Food Chem Toxicol 88:87–99
Lindbäck T, Fagerlund A, Rødland MS et al (2004) Characterization of the Bacillus cereus Nhe enterotoxin. Microbiology 150:3959–3967
Lisdawati V, Puspandari N, Rif’ati L et al (2015) Molecular epidemiology study of Mycobacterium tuberculosis and its susceptibility to anti-tuberculosis drugs in Indonesia. BMC Infect Dis 15:366
Lund T, Granum PE (1996) Characterisation of a non-haemolytic enterotoxin complex from Bacillus cereus isolated after a foodborne outbreak. FEMS Microbiol Lett 141:151–156
McIntyre L, Bernard K, Beniac D et al (2008) Identification of Bacillus cereus group species associated with food poisoning outbreaks in British Columbia, Canada. Appl Environ Microbiol 74(23):7451–7453
Moravek M, Wegscheider M et al (2004) Colony immunoblot assay for the detection of hemolysin BL enterotoxin producing Bacillus cereus. FEMS Microbiol Lett 238(1):107–113
Okinaka RT, Keim P (2016) The phylogeny of Bacillus cereus sensu lato. Microbiol Spectr 4(1): TBS-0012-2012
Samapundo S, Heyndrickx M et al (2011) Incidence, diversity and toxin gene characteristics of Bacillus cereus group strains isolated from food products marketed in Belgium. Int J Food Microbiol 150(1):34–41
Sorokin A, Candelon B, Guilloux K et al (2006) Multiple-locus sequence typing analysis of Bacillus cereus and Bacillus thuringiensis reveals separate clustering and a distinct population structure of psychrotrophic strains. Appl Environ Microbiol 72:1569–1578
Swiecicka I, Van der Auwera GA, Mahillon J (2006) Hemolytic and nonhemolytic enterotoxin genes are broadly distributed among Bacillus thuringiensis isolated from wild mammals. Microb Ecol 52(3):544–551
Virginie C, Xiaojin L, Laurence D et al (2015) Is Cytotoxin K from Bacillus cereus a bona fide enterotoxin? Int J Food Microbiol 211:79–85
Wehrle E, Moravek M, Dietrich R et al (2009) Comparison of multiplex PCR, enzyme immunoassay and cell culture methods for the detection of enterotoxinogenic Bacillus cereus. J Microbiol Methods 78(3):265–270
Zhou G, Zheng D, Dou L et al (2010) Occurrence of psychrotolerant Bacillus cereus group strains in ice creams. Int J Food Microbiol 137:143–146
Zhu Y, Chen P, Bao Y et al (2016) Complete genome sequence and transcriptomic analysis of a novel marine strain Bacillus weihaiensis reveals the mechanism of brown algae degradation. Sci Rep 30(6):38248
Acknowledgements
This study was supported by grants from Key project of Anhui Science and Technology Department (1408085QC59), Key project of Anhui Education Department (KJ2016A849), Anhui Provincial Key Lab of the Conservation and Exploitation of Biological Resources, Innovation Team of Scientific Research Platform in Anhui Universities, Chinese National Natural Science Foundation of China (No.31500418), Jiangsu Province Natural Science Foundation of China (BK200140261).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Jorge Membrillo-Hernández.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cai, Y., Huang, T., Xu, Y. et al. Genetic and genomic diversity of NheABC locus from Bacillus strains. Arch Microbiol 199, 775–785 (2017). https://doi.org/10.1007/s00203-017-1350-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-017-1350-9