
Abstract
Aims/hypothesis
Recurrent hypoglycaemia is primarily caused by repeated over-administration of insulin to patients with diabetes. Although cognition is impaired during hypoglycaemia, restoration of euglycaemia after recurrent hypoglycaemia is associated with improved hippocampally mediated memory. Recurrent hypoglycaemia alters glucocorticoid secretion in response to hypoglycaemia; glucocorticoids are well established to regulate hippocampal processes, suggesting a possible mechanism for recurrent hypoglycaemia modulation of subsequent cognition. We tested the hypothesis that glucocorticoids within the dorsal hippocampus might mediate the impact of recurrent hypoglycaemia on hippocampal cognitive processes.
Methods
We characterised changes in the dorsal hippocampus at several time points to identify specific mechanisms affected by recurrent hypoglycaemia, using a well-validated 3 day model of recurrent hypoglycaemia either alone or with intrahippocampal delivery of glucocorticoid (mifepristone) and mineralocorticoid (spironolactone) receptor antagonists prior to each hypoglycaemic episode.
Results
Recurrent hypoglycaemia enhanced learning and also increased hippocampal expression of glucocorticoid receptors, serum/glucocorticoid-regulated kinase 1, cyclic AMP response element binding (CREB) phosphorylation, and plasma membrane levels of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartic acid (NMDA) receptors. Both hippocampus-dependent memory enhancement and the molecular changes were reversed by glucocorticoid receptor antagonist treatment.
Conclusions/interpretation
These results indicate that increased glucocorticoid signalling during recurrent hypoglycaemia produces several changes in the dorsal hippocampus that are conducive to enhanced hippocampus-dependent contextual learning. These changes appear to be adaptive, and in addition to supporting cognition may reduce damage otherwise caused by repeated exposure to severe hypoglycaemia.
Similar content being viewed by others

Introduction
Hypoglycaemia occurs in patients with either type 1 or type 2 diabetes, at a rate of around 43 bouts per patient per year in type 1 diabetes and 16 bouts per patient per year in type 2 [1]. The bouts vary in length, but can last for hours if they are nocturnal [2] and/or exacerbated by other lifestyle factors such as exercise [3]. Reports vary, but around 5–10% of individuals with type 1 diabetes may die from hypoglycaemic complications [4, 5].
During hypoglycaemia, cognitive processing is highly diminished because of the impaired supply of glucose to the brain [6]. Moreover, studies in both animals and humans suggest that adaptation occurs in type 1 diabetes to alter cognitive function during successive hypoglycaemic bouts [7, 8]. Following nocturnal hypoglycaemia, cognitive function in patients with type 1 diabetes is better preserved during subsequent hypoglycaemia [9], while short-term memory, reaction times and P300 amplitudes and latencies are improved or maintained following recurrent hypoglycaemia compared with those measured after an initial antecedent hypoglycaemic bout [6, 10]. In rodents, this has been extended to show that, following recovery from recurrent hypoglycaemia, euglycaemic working memory is significantly improved compared with animals that did not undergo hypoglycaemia [8].
Relatively few studies have examined the impact of recurrent hypoglycaemia on cognitive function in humans, and the results are often potentially confounded by other effects of diabetes that are generally deleterious; the mechanisms underlying adaptive responses to recurrent hypoglycaemia that result in preservation and/or enhancement of cognition are not well understood. Here, we investigated a potential role for glucocorticoids in mediating hippocampal responses to recurrent hypoglycaemia that are responsible for procognitive effects on behaviour in rats. We used a rat model of recurrent hypoglycaemia that we have previously validated [8] in order to avoid any impact of any underlying disease state.
Hypoglycaemia is stressful, activating the hypothalamic–pituitary–adrenal (HPA) axis. HPA-stimulated glucocorticoid release plays a major role in maintaining systemic blood glucose levels [11]. Glucocorticoids also have profound effects on the brain. Once released from the adrenals, glucocorticoids reach the hippocampus within minutes (peaking at 8–10 min [12]). Although both mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs) are present throughout the brain, the hippocampus has the greatest brain level of receptor co-localisation [13].
Recurrent hypoglycaemia affects the hippocampus directly, including effects on glucocorticoid signalling. Adolescent recurrent hypoglycaemia chronically reduces hypothalamic corticotrophin releasing hormone and GRs, but elevates both GRs and MRs in the hippocampus [14]. A similar regional contrast is seen with regard to cell death: glucose-sensing regions of the hypothalamus undergo apoptosis [15] and suppression [16], but the hippocampus adapts to hypoglycaemia, maintains synaptic transmission [17], increases glucose availability [8] and increases glutamatergic signalling during hypoglycaemia [18]. These changes may underlie altered cognitive performance: brain glucocorticoid signalling transduces the impact of recurrent hypoglycaemia on hypothalamic glucose-sensing and impaired awareness of hypoglycaemia [19], supporting this mechanism as a plausible candidate to mediate the neural impact of recurrent hypoglycaemia in the hippocampus.
The dorsal hippocampus is integral to both learning and spatial navigation, including memory for contextual, but not cued, conditioned fear [20, 21]. N-Methyl-d-aspartic acid (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor activation triggers calcium influx [22], with downstream effects including activation of cyclic AMP response element binding (CREB) protein. Phosphorylation of CREB at Ser133 (to produce pCREB) is considered crucial for hippocampal memory formation, including contextual fear learning [23]. Glucocorticoids regulate several hippocampal molecular memory processes. At physiological levels, glucocorticoids in the dorsal hippocampus improve learning and memory in a time- and dose-dependent manner [24]. Growing evidence supports a role for serum/glucocorticoid-inducible kinase 1 (SGK1), an early gene product of nuclear GR and/or MR activation [25, 26], in mediating the cognitive enhancing effects of glucocorticoids. SGK1 mediates performance in water maze and contextual fear tasks [27, 28]. Activated SGK1 has been shown to increase CREB Ser133 phosphorylation [29], NMDA receptor (NMDAR) 2B expression [30] and NMDA/AMPA receptor trafficking [31, 32]. Peripherally, SGK1 has been implicated in the pathological consequences of both type 1 and type 2 diabetes [33, 34]. Overall, we identified SGK1 as a potential candidate transducer for the impact of glucocorticoids and/or hypoglycaemia on the dorsal hippocampus.
We took molecular measures at three time points (Fig. 1) to characterise the impact of recurrent hypoglycaemia on several potential molecular targets of interest capable of influencing cognition. Our hypothesis was that recurrent hypoglycaemia would alter hippocampal physiology at baseline, improve performance on a contextual fear task and perhaps show an interaction with testing in terms of molecular changes; these hypotheses were confirmed. In addition, many behavioural and molecular consequences of recurrent hypoglycaemia were shown to be dependent on glucocorticoid signalling in the dorsal hippocampus, providing the first mechanistic explanation for the impact of recurrent hypoglycaemia on memory processing.
Methods
Animals
Procedures were approved by the University Institutional Animal Care and Use Committee and adhered to standards set forth by the Guide for the Care and Use of Laboratory Animals. Male Sprague Dawley rats (Crl:SD outbred strain, Charles River, Wilmington, MA, USA) arrived at 11 weeks old and were given ad libitum food and water except during hypoglycaemia (see the Recurrent hypoglycaemia section below). Rats were maintained on a 12/12 light/dark schedule, with lights switched on at 07:00 hours. Rats rested in the facility for 1 week before any experiments took place. Prior to surgery, rats were housed two to a cage; following surgery rats were single-housed. All procedures began between 09:00 hours and 12:00 hours; hypoglycaemia was terminated by 15:00 hours.
Infusion treatments
The vehicle was artificial extracellular fluid (aECF; [35]) with 1.25 mmol/l glucose. For animals receiving glucocorticoid antagonist treatment, the MR and GR antagonists spironolactone and mifepristone (Sigma-Aldrich, St Louis, MO, USA), respectively, were prepared together in aECF with 0.01% ethanol to a final concentration of 5 ng/0.5 μl [36] and given at 0.5 μl per side at 0.125 μl/min immediately prior to hypoglycaemia or saline (154 mmol/l NaCl) administration.
Experimental groups
All groups underwent the same surgical and recurrent hypoglycaemia schedule, but group experiences varied beginning 24 h after the third bout of hypoglycaemia (Fig. 1). Animals were randomly assigned to one of four conditions: saline + aECF, saline + glucocorticoid (GC) antagonist, recurrent hypoglycaemia (RH) + aECF, or RH + GC antagonist. GC antagonist conditions were randomly assigned by co-authors not involved in the behavioural testing processes; the experimenter was blinded to all animal conditions during contextual fear testing and scoring. Between two and four separate cohort replicates were created and run for each of the four groups listed below. Group sizes were based on previous studies that examined behavioural and metabolic effects of recurrent hypoglycaemia [8].

Timeline for testing and tissue collection. Experimental procedures were identical for all groups until the end of the third day of hypoglycaemia. The RH-only group had tissues collected 24 h after their final bout of recurrent hypoglycaemia. The learning group received shock–context pairing in the training chamber, with tissues collected 4 h after training. The retrieval group underwent conditioned fear training with shock–context pairing; their post-fear retrieval behaviour was measured 24 h after training, and tissues were collected immediately afterwards. The context group was exposed to the context with no shock pairing, and tissues were collected immediately after the behaviour. Hippo., hippocampus
RH-only group
For the RH group, tissues were collected 24 h after the third bout of hypoglycaemia. Group numbers were as follows: saline + aECF, n = 7; saline + GC antagonist, n = 6; RH + aECF, n = 7; RH + GC antagonist, n = 4.
Learning group
Contextual fear training was applied, and the animals were euthanised 4 h after training. Home-cage controls were run in parallel with this group to establish baseline levels of protein expression. These rats were regularly handled but did not undergo surgery, infusions or recurrent hypoglycaemia. Group numbers were as follows: saline + aECF, n = 5; RH + aECF, n = 8; RH + GC antagonist, n = 7; home-cage, n = 4.
Retrieval group
The animals underwent contextual fear training and testing, and were euthanised immediately. Group numbers were as follows: saline + aECF, n = 11; saline + GC antagonist, n = 5; RH + aECF, n = 11; RH + GC antagonist, n = 10.
Context group
This group served as a behavioural control for the retrieval group. They were exposed to the context training chamber but did not receive foot shocks. Conditions were otherwise identical to the retrieval group, to permit controlling for any behavioural and molecular changes due to the novel environment. Group numbers were as follows: saline + aECF, n = 5; saline + GC antagonist, n = 8; RH + aECF, n = 6; RH + GC antagonist, n = 6.
Cannula implantation
Rats were anaesthetised with 5% isoflurane. Standard sterile stereotaxic methods were used to implant microinjection cannulae into the dorsal hippocampus at coordinates (in mm, relative to the bregma) of −5.6 posterior, ±5.0 lateral and −3.3 ventral. Rimadyl (5 mg/kg body weight, BioServ, Flemington, NJ, USA) was given postsurgically and daily for 3 days thereafter. Animals were given 7 days of recovery, with extensive handling each day. Cannula placement was confirmed at tissue extraction. If the probe was misplaced, behavioural and biochemistry results for that animal were excluded from all analyses. Four rats were removed from the study due to misplaced probes.
Recurrent hypoglycaemia
Hypoglycaemia was induced for 3 h once daily for three consecutive days [8]. Rats received 10 U/kg i.p. insulin on the first day; subsequent doses were titrated (4–9 U/kg) to ensure blood glucose levels between 2.2 and 2.8 mmol/l as the rats sensitised to insulin over the 3 day period (see Table 1 for average values). Control animals received volume-matched i.p. saline injections. Blood was collected by tail-vein prick 1 h after insulin administration for blood glucose determination (OneTouch Ultra meter, LifeScan, Wayne, PA, USA) and corticosterone levels (Yale Medical Labs, New Haven, CT, USA). Rats that did not experience blood glucose levels below 2.8 mmol/l for 3 days were excluded from the study (n = 3).
Contextual fear conditioning
Behavioural training was begun 24 h after the last hypoglycaemic bout. Contextual fear measures were adapted [37] to test hippocampus-dependent spatial memory. Rats were placed in a chamber with distinct spatial cues and allowed to explore freely for 60 s prior to presentation of a tone (80 db) for 10 s followed immediately by a 5 s, 0.8 mA foot shock. This tone–shock pairing was repeated three times with 1 min intertrial intervals. Rats were returned to their home cage until testing 24 h later, when they were returned to the same chamber. Memory for association between context and foot shock was assessed by time spent freezing without the tone, defined as a complete absence of movement.
Western blotting
The dorsal hippocampus was removed and homogenised with 300 μl of homogenisation buffer containing phosphate inhibitor (1:500), from which 30 μl of homogenate was removed and combined with RIPA buffer containing protease and phosphatase inhibitors. The initial homogenate was processed for plasma membrane isolation as per the manufacturer’s instructions (BioVision, Milpitas, CA, USA). Protein was loaded into SDS PAGE 10% gels (BioRad). Gels were transferred to PVDF membrane (Thermo Scientific, Waltham, MA, USA). Primary antibodies were left overnight at 4°C for GR (1:1000, Cell Signaling, Beverly, MA, USA), AMPA receptor (AMPAR, subtype 1) (1:1000, Cell Signaling), actin (1:10,000, Sigma-Aldrich, St. Louis, MO, USA), NMDAR 2B (1:1000, Cell Signaling), SGK1 (1:750, Millipore, Billerica, MA, USA), pCREB (1:500, Abcam, Cambridge, MA, USA) and CREB (1:750, Abcam). Anti-rabbit and anti-mouse secondary antibodies were used at 1:20,000 in TBST and then with tertiary streptavidin–horseradish peroxidase (Cell Signaling) for 1 h. SuperSignal West Pico chemiluminescent (Thermo Scientific) was applied. Bands were analysed using Image J software version 1.45 s (NIH, http://imagej.nih.gov/ij/).
Data analysis
Statistical analyses used one- , two- and three-way ANOVAs via GraphPad Prism 6 (La Jolla, CA, USA) and SPSS version 17.0 (http://www.ibm.com/analytics/us/en/technology/spss/), α = 0.05, with Bonferroni multiple comparison post hoc tests as needed. Specific planned comparisons were made between (1) RH and saline-treated animals, and (2) GC antagonist conditions. Data points 3 SD or more away from the mean were excluded. In some cases, this caused minor variation in n between measures.
Results
GC antagonism did not affect induction of hypoglycaemia, but altered corticosterone release when combined with recurrent hypoglycaemia
Induction of hypoglycaemia successfully lowered blood glucose to target levels (F 1,75 = 1381, p < 0.0001; Table 1), unaffected by either day of hypoglycaemia (F 2,75 = 2.003, p > 0.05) or GC antagonism (F 1,75 = 1.604, p > 0.05).
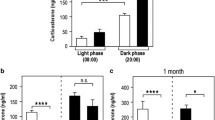
There were significant group differences in peripheral corticosterone levels (Table 1). As expected, recurrent hypoglycaemia increased corticosterone concentrations (F 1,63 = 120.019, p < 0.0001), and the increase in corticosterone with recurrent hypoglycaemia decreased across the 3 days of recurrent hypoglycaemia (main effect of day F 2,63 = 11.256, p < 0.001; interaction between recurrent hypoglycaemia and day F 2,63 = 5.427, p < 0.01). GC antagonism potentiated the increased corticosterone levels in RH animals (interaction between RH and GC antagonism, F 1,63 = 5.007, p < 0.05).
Insulin administration decreased across the 3 days of RH (Table 1; F 2,36 = 61.97, p < 0.0001). Post hoc analysis showed a difference between aECF and GC antagonism on the third day of RH: antagonist-infused rats required less insulin compared with vehicle (p < 0.05).
Glucocorticoid antagonism during hypoglycaemia prevented recurrent hypoglycaemia-induced enhancement of hippocampus-dependent contextual memory
In the retrieval group, there was a significant interaction (F 1,32 = 6.827, p < 0.05; Fig. 2a) between recurrent hypoglycaemia and GC antagonism in affecting freezing in response to context. Recurrent hypoglycaemia significantly increased freezing, indicating improved memory, and this effect was not seen in animals receiving GC antagonism.

Percentage time freezing in the contextual fear task for the retrieval group (a) and the context-only group (b), which were exposed to the same context but not shocked. In the retrieval group (a), there was a significant interaction in which recurrent hypoglycaemia significantly increased the time spent freezing, an effect attenuated by the addition of GC antagonists. In the context-only group (b), there were no differences in freezing. Black bars, aECF-infused rats; white bars, GC antagonist-infused rats. *p < 0.05 compared with saline + aECF group; § p < 0.05 compared with RH + GC antagonist group
Rats that were exposed to context without foot shock showed no group differences in freezing: no effect of RH (F 1,21 = 1.390, p > 0.05), GC antagonism (F 1,21 = 0.0803, p > 0.05) or their interaction (F 1,21 = 0.9805, p > 0.05; Fig. 2b).
Recurrent hypoglycaemia alone altered key protein levels in the dorsal hippocampus; glucocorticoid antagonism attenuated some of the increases
In the RH-only group, recurrent hypoglycaemia increased dorsal hippocampal GRs (main effect of recurrent hypoglycaemia F 1,16 = 6.375, p < 0.05; Fig. 3b) and SGK1 (F 1,17 = 6.350, p < 0.05; Fig. 3a), but neither measure was affected by GC antagonism, nor was there an interaction between recurrent hypoglycaemia and antagonism.

Recurrent hypoglycaemia increased total SGK1 (a) and GR (b) in the dorsal hippocampus regardless of GC antagonism. There was a significant interaction between RH and GC antagonism as modulators of plasma membrane (PM) AMPAR (c) and plasma membrane NMDAR (d). No significant changes were found in total AMPAR (e) or total NMDAR (f). Black bars, aECF-infused rats; white bars, GC antagonist-infused rats. *p < 0.05 compared with saline + aECF group; † p < 0.05 compared with saline + GC antagonist group; § p < 0.05 compared with RH + GC antagonist group. AU, absorbance units
Plasma membrane NMDAR measurements showed a significant interaction for RH × GC antagonism (F 1,14 = 15.68, p < 0.05; Fig. 3d),but there was no main effect of RH or GC antagonism. Planned comparisons showed that recurrent hypoglycaemia increased plasma membrane NMDAR and that this increase was reversed in animals in the RH + GC antagonist group. Unexpectedly, additional post hoc comparisons indicated that plasma membrane NMDAR levels were also increased in animals receiving GC antagonism only, compared with animals receiving either saline + aECF or RH + GC antagonism.
There was a significant interaction between RH and GC antagonism (F 1,16 = 48.05, p < 0.0001; Fig. 3c), but no effect of RH or GC antagonism. Planned comparisons confirmed that this reflected an increase in plasma membrane APMAR that was caused by recurrent hypoglycaemia and attenuated by GC antagonism. As with the NMDAR measures, GC antagonism alone unexpectedly increased AMPAR levels compared with animals receiving either saline + aECF or RH + GC antagonism.
Total hippocampal NMDAR and AMPAR were also measured. There was a significant interaction (F 1,18 = 4.48, p < 0.05; Fig. 3e, f) for total NMDAR levels. There were no main effects of either RH or GC antagonist treatment. Total AMPAR levels were not significantly affected by any treatments.
Recurrent hypoglycaemia increases hippocampal SGK1 and pCREB after task encoding, an effect that is reversed by GC antagonism
In the learning group, 4 h after training in the contextual fear task there were significant one-way ANOVA effects for treatment indicating changes in dorsal hippocampal levels of SGK1 (F 3,23 = 5.078, p < 0.01; Fig. 4a, b) and pCREB (F 3,23 = 3.170, p < 0.05). Both SGK1 and pCREB levels were significantly increased in the RH + aECF group compared with saline + aECF and significantly attenuated by the addition of GC antagonist. There were no differences in plasma membrane or total NMDAR or AMPAR levels (data not shown).

Recurrent hypoglycaemia increased hippocampal SGK1 (a) and pCREB (b) 4 h after contextual fear training; GC antagonism reversed this increase. Home-cage controls did not vary from saline or RH + GC antagonist groups. *p < 0.05 compared with saline + aECF group; § p < 0.05 compared with RH + GC antagonist group. AU, absorbance units
Recurrent hypoglycaemia increases, and hippocampal gluococorticoid antagonism restores, pCREB following retrieval while contextual exposure alone has no effect
In the retrieval group, pCREB was elevated in RH-aECF animals; this increase was reversed by GC antagonism (F 1,28 = 4.464, p < 0.05 for the RH-antagonism interaction; Fig. 5a, b). No group differences were seen in SGK1. In the context group, exposure to the context chamber alone did not produce any changes in protein measures, nor were any group differences seen in this group (Fig. 6a, b). There were no differences in plasma membrane or total NMDAR or AMPAR levels (data not shown).

Relative levels of SGK1 (a) and pCREB (b) in the behavioural group immediately after behavioural testing. There were no differences across conditions for SGK1. Recurrent hypoglycaemia increased pCREB, an effect attenuated by the addition of GC antagonists. Black bars, aECF-infused rats; white bars, GC antagonist-infused rats. *p < 0.05 compared with saline + aECF group; § p < 0.05 compared with RH + GC antagonist group. AU, absorbance units

Relative levels of SGK1 (a) and pCREB (b) in the dorsal hippocampus of the context-only group immediately following behavioural testing. There were no differences across conditions. Black bars, aECF-infused rats; white bars, GC antagonist-infused rats. AU, absorbance units
Discussion
The hypothesis that glucocorticoid actions in the dorsal hippocampus mediate the effects of recurrent hypoglycaemia on hippocampal learning and memory was supported. Recurrent hypoglycaemia enhanced subsequent contextual fear memory (tested at euglycaemia, Fig. 2a), extending, and consistent with, prior work using spatial working memory [8, 38]. This enhancement was blocked by intrahippocampal glucocorticoid antagonism during each hypoglycaemic episode (Fig. 2a). Several glucocorticoid-dependent molecular mechanisms contributing to this behavioural effect were identified. Twenty-four hours after the third daily bout of hypoglycaemia, recurrent hypoglycaemia increased hippocampal GR, SGK1 and plasma membrane AMPAR and NMDAR, with the increase in plasma membrane AMPAR and NMDAR being dependent on glucocorticoids (Fig. 3a–f). Additional glucocorticoid-dependent effects of recurrent hypoglycaemia were seen after both formation of a contextual fear memory and retrieval of that memory: increased hippocampal SGK1 and pCREB after training (Fig. 4a, b), and increased pCREB after retrieval (Fig. 5b), were all shown to be dependent on hippocampal glucocorticoid signalling during recurrent hypoglycaemia. Because exposure to a novel context alone did not result in commensurate changes, regardless of treatment, these data support a glucocorticoid-dependent role for recurrent hypoglycaemia in enhancing hippocampal long-term memory processes via increased pCREB and SGK1 activity.
Hippocampal glucocorticoid antagonism during recurrent hypoglycaemia also potentiated systemic release of the counterregulatory hormone corticosterone. Several studies have demonstrated that recurrent hypoglycaemia attenuates corticosterone levels in plasma during subsequent hypoglycaemic episodes, a key element of eventual counterregulatory failure after recurrent hypoglycaemia [39]. Animals that received the GR/MR antagonists still experienced a decline in peripheral corticosterone levels due to recurrent hypoglycaemia across days, but their corticosterone levels were higher than those of animals in the RH–aECF group (Table 1). This was probably due to impaired hippocampal feedback termination of the HPA response, allowing for increased and/or prolonged secretion of corticosterone. Because glucocorticoid antagonist treatments were delivered specifically to the hippocampus, initiation of a counterregulatory response by the hypothalamus would probably remain unaffected.
Prior to any behavioural task, recurrent hypoglycaemia increased hippocampal SGK1 and glucocorticoid receptors, both in a non-glucocorticoid-dependent manner (Fig. 3a, b). This finding was again surprising, as both proteins are under genomic regulation by glucocorticoids [25, 26]. Increased hippocampal glucocorticoid receptors would be expected to lead to a more rapid subsequent cessation of counterregulatory hormone release, and merits further study in terms of the impaired counterregulation seen after recurrent hypoglycaemia. SGK1 may, perhaps, require additional stimuli (specifically, cognitive engagement) in order for glucocorticoid sensitivity of changes in response to recurrent hypoglycaemia to become more apparent: in the learning group 4 h after training, SGK1 levels were again elevated due to recurrent hypoglycaemia (Fig. 4a), but in contrast to the baseline changes this increase was glucocorticoid-dependent. SGK1 is involved in hippocampus-dependent forms of memory [27, 28, 32], and may be partially responsible for the procognitive effects of recurrent hypoglycaemia.
CREB phosphorylation at Ser133 is important for hippocampally mediated spatial learning [23]. Here, hippocampal pCREB was increased by recurrent hypoglycaemia, and attenuated by the addition of GC antagonism, 4 h after training (Fig. 4b), a critical period in pCREB-related consolidation events [40, 41]. At the same time point, hippocampal SGK1 was also increased by recurrent hypoglycaemia via glucocorticoid signalling (Fig. 4a). Limited work has been done to characterise SGK1 in the hippocampus [27, 32]; chronic upregulation of SGK1 enhances, while knockdown decreases, contextual fear learning [28], consistent with our data. Although SGK1 can increase pCREB [29], the fact that we saw an increase in pCREB after retrieval without any change in SGK1 suggests that the glucocorticoid-dependent effects of recurrent hypoglycaemia to increase hippocampus-dependent long-term spatial learning may also be due to direct glucocorticoid activation of CREB during retrieval, independent of SGK1. Because animals exposed only to the contextual chamber (the context group) did not have any changes in hippocampal pCREB (Fig. 6b), increased pCREB levels after learning and retrieval can be attributed to recurrent hypoglycaemia, and were glucocorticoid-dependent. Animals in the context group, as expected, showed very little freezing behaviour (Fig. 2b) and no effect of any treatment on freezing, validating recurrent hypoglycaemia and glucocorticoid antagonism as the sources of behavioural and protein group differences observed.
Also prior to any behavioural training or testing, both recurrent hypoglycaemia and glucocorticoid antagonists increased hippocampal plasma membrane levels of AMPAR and NMDA (Fig. 3c, d). This may explain previous findings of increased synaptic activity [17] and improved working memory [8] in rats after recurrent hypoglycaemia. The finding that glucocorticoid antagonists increased hippocampal glutamate receptor levels was contrary to expectation, given that glucocorticoids increase both receptors at the plasma membrane [42, 43]. These apparently paradoxical results may be due to experimental differences: previous measurements of the effects of glucocorticoids on plasma membrane levels of AMPAR and NMDAR were made after acute MR/GR manipulation, whereas the current study measured protein levels 24 h after the last of three antagonist doses. We speculate that GR/MR antagonism over the course of 3 days may have prompted a compensatory increase in glutamate transmission capacity, similar to that seen for glucose transport in response to recurrent hypoglycaemia. Glucocorticoid antagonists alone did not affect either SGK1 or pCREB, our primary molecular measures, both of which were significantly affected by recurrent hypoglycaemia and attenuated by glucocorticoid antagonism. The increase in plasma membrane glutamate receptors, seen after antagonist administration, was not sufficient alone to affect behavioural outcomes: although follow-up studies on glutamate transmission after recurrent hypoglycaemia would be of interest, we interpret our results to more strongly implicate SGK1 working through pCREB as a pathway of interest, and as warranting additional research into pCREB’s downstream effects on brain derived neurotrophic factor [44], activity-regulated cytoskeleton-associated (Arc/Arg3.1) protein [45], c-fos [45] and/or early growth response-1 protein (EGR1) [45] as further downstream targets of the effects of recurrent hypoglycaemia on hippocampal cognition.
Neuronal responses to hypoglycaemia will vary with the severity of hypoglycaemia. Here, we used a well-established moderate recurrent hypoglycaemia method, but more severe hypoglycaemia is likely to further elevate glucocorticoid levels, with unknown long-term consequences. It is important to note that these results were obtained in non-diabetic animals, and hence are unconfounded by a diabetic disease state. Following recurrent hypoglycaemia in a hippocampal-dependent working memory task, studies have demonstrated identical cognitive enhancement in type 1 diabetic and non-diabetic rat models [8], suggesting that similar behavioural and physiological changes would occur in rats with type 1 diabetes using this paradigm; however, a possible limitation is that we did not study the interaction of recurrent hypoglycaemia with diabetes. Many of the changes induced by recurrent hypoglycaemia in the hippocampus were attenuated by glucocorticoid antagonism; this introduces a potential clinical complication in terms of how insulin treatments should be managed in clinical populations. Anti-glucocorticoid treatments may interact with hypoglycaemic medications, including insulin, to minimise adaptive responses and potentially impair susceptibility to future hypoglycaemic bouts. Further work using clinically relevant routes of glucocorticoid antagonist administration might give insight into this, and also into whether a therapeutic modulation of glucocorticoid signalling could be beneficial in alleviating the cognitive deficits that occur during hypoglycaemia, a critical issue in patients with diabetes [46].
Abbreviations
- aECF:
-
Artificial extracellular fluid
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- CREB:
-
Cyclic AMP response element binding
- GC:
-
Glucocorticoid (treatment group)
- GR:
-
Glucocorticoid receptor
- HPA:
-
Hypothalamic–pituitary–adrenal
- MR:
-
Mineralocorticoid receptor
- NMDA:
-
N-Methyl-d-aspartic acid
- NMDAR:
-
N-Methyl-d-aspartic acid receptor
- pCREB:
-
Phosphorylated CREB
- RH:
-
Recurrent hypoglycaemia (treatment group)
- SGK1:
-
Serum/glucocorticoid-inducible kinase-1
References
Donnelly LA, Morris AD, Frier BM et al (2005) Frequency and predictors of hypoglycaemia in type 1 and insulin-treated type 2 diabetes: a population-based study. Diabet Med : J Br Diabet Assoc 22:749–755
Buckingham B, Wilson DM, Lecher T, Hanas R, Kaiserman K, Cameron F (2008) Duration of nocturnal hypoglycemia before seizures. Diabetes Care 31:2110–2112
Graveling AJ, Frier BM (2010) Risks of marathon running and hypoglycaemia in type 1 diabetes. Diabet Med : J Br Diabet Assoc 27:585–588
Deckert T, Poulsen JE, Larsen M (1978) Prognosis of diabetics with diabetes onset before the age of thirty-one. I. Survival, causes of death, and complications. Diabetologia 14:363–370
Skrivarhaug T, Bangstad HJ, Stene LC, Sandvik L, Hanssen KF, Joner G (2006) Long-term mortality in a nationwide cohort of childhood-onset type 1 diabetic patients in Norway. Diabetologia 49:298–305
Fruehwald-Schultes B, Born J, Kern W, Peters A, Fehm HL (2000) Adaptation of cognitive function to hypoglycemia in healthy men. Diabetes Care 23:1059–1066
Lobmann R, Smid HG, Pottag G, Wagner K, Heinze HJ, Lehnert H (2000) Impairment and recovery of elementary cognitive function induced by hypoglycemia in type-1 diabetic patients and healthy controls. J Clin Endocrinol Metab 85:2758–2766
McNay EC, Sherwin RS (2004) Effect of recurrent hypoglycemia on spatial cognition and cognitive metabolism in normal and diabetic rats. Diabetes 53:418–425
Fanelli CG, Paramore DS, Hershey T et al (1998) Impact of nocturnal hypoglycemia on hypoglycemic cognitive dysfunction in type 1 diabetes. Diabetes 47:1920–1927
Schultes B, Kern W, Oltmanns K et al (2005) Differential adaptation of neurocognitive brain functions to recurrent hypoglycemia in healthy men. Psychoneuroendocrinology 30:149–161
Lundgren M, Buren J, Ruge T, Myrnas T, Eriksson JW (2004) Glucocorticoids down-regulate glucose uptake capacity and insulin-signaling proteins in omental but not subcutaneous human adipocytes. J Clin Endocrinol Metab 89:2989–2997
Barbaccia ML, Serra M, Purdy RH, Biggio G (2001) Stress and neuroactive steroids. Int Rev Neurobiol 46:243–272
Van Eekelen JA, Jiang W, De Kloet ER, Bohn MC (1988) Distribution of the mineralocorticoid and the glucocorticoid receptor mRNAs in the rat hippocampus. J Neurosci Res 21:88–94
Lehmann AE, Ennis K, Georgieff MK, Rao R, Tran PV (2011) Evidence for a hyporesponsive limbic-hypothalamic-pituitary-adrenal axis following early-life repetitive hypoglycemia in adult male rats. Am J Physiol Regul, Integr Comp Physiol 301:R484–R490
Tkacs NC, Dunn-Meynell AA, Levin BE (2000) Presumed apoptosis and reduced arcuate nucleus neuropeptide Y and pro-opiomelanocortin mRNA in non-coma hypoglycemia. Diabetes 49:820–826
Evans SB, Wilkinson CW, Bentson K, Gronbeck P, Zavosh A, Figlewicz DP (2001) PVN activation is suppressed by repeated hypoglycemia but not antecedent corticosterone in the rat. Am J Physiol Regul, Integr Comp Physiol 281:R1426–R1436
Sakurai T, Yang B, Takata T, Yokono K (2002) Synaptic adaptation to repeated hypoglycemia depends on the utilization of monocarboxylates in guinea pig hippocampal slices. Diabetes 51:430–438
Sandberg M, Butcher SP, Hagberg H (1986) Extracellular overflow of neuroactive amino acids during severe insulin-induced hypoglycemia: in vivo dialysis of the rat hippocampus. J Neurochem 47:178–184
Kale AY, Paranjape SA, Briski KP (2006) I.c.v. administration of the nonsteroidal glucocorticoid receptor antagonist, CP-472555, prevents exacerbated hypoglycemia during repeated insulin administration. Neuroscience 140:555–565
Broadbent NJ, Squire LR, Clark RE (2004) Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci U S A 101:14515–14520
Hunsaker MR, Kesner RP (2008) Dissociations across the dorsal-ventral axis of CA3 and CA1 for encoding and retrieval of contextual and auditory-cued fear. Neurobiol Learn Mem 89:61–69
Miyamoto E (2006) Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci 100:433–442
Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR (1998) Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci 1:595–601
Joels M (2006) Corticosteroid effects in the brain: U-shape it. Trends Pharmacol Sci 27:244–250
Chen SY, Bhargava A, Mastroberardino L et al (1999) Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A 96:2514–2519
Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL (1993) Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol 13:2031–2040
Tsai KJ, Chen SK, Ma YL, Hsu WL, Lee EHY (2002) Sgk, a primary glucocorticoid-induced gene, facilitates memory consolidation of spatial learning in rats. Proc Natl Acad Sci U S A 99:3990–3995
Lee CT, Ma YL, Lee EH (2007) Serum- and glucocorticoid-inducible kinase1 enhances contextual fear memory formation through down-regulation of the expression of Hes5. J Neurochem 100:1531–1542
David S, Kalb RG (2005) Serum/glucocorticoid-inducible kinase can phosphorylate the cyclic AMP response element binding protein, CREB. FEBS Lett 579:1534–1538
Tai DJ, Su CC, Ma YL, Lee EH (2009) SGK1 phosphorylation of IkappaB kinase alpha and p300 Up-regulates NF-kappaB activity and increases N-methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem 284:4073–4089
Yuen EY, Liu W, Karatsoreos IN et al (2011) Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry 16:156–170
Lee EH, Hsu WL, Ma YL, Lee PJ, Chao CC (2003) Enrichment enhances the expression of sgk, a glucocorticoid-induced gene, and facilitates spatial learning through glutamate AMPA receptor mediation. Eur J Neurosci 18:2842–2852
Lang F, Gorlach A, Vallon V (2009) Targeting SGK1 in diabetes. Expert Opin Ther Targets 13:1303–1311
Schwab M, Lupescu A, Mota M et al (2008) Association of SGK1 gene polymorphisms with type 2 diabetes. Cell Physiol Biochem : Int J Exp Cell Physiol, Biochem Pharmacol 21:151–160
McNay EC, Gold PE (1999) Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J Neurochem 72:785–790
van Haarst AD, Oitzl MS, de Kloet ER (1997) Facilitation of feedback inhibition through blockade of glucocorticoid receptors in the hippocampus. Neurochem Res 22:1323–1328
Johansen JP, Cain CK, Ostroff LE, LeDoux JE (2011) Molecular mechanisms of fear learning and memory. Cell 147:509–524
McNay EC, Williamson A, McCrimmon RJ, Sherwin RS (2006) Cognitive and neural hippocampal effects of long-term moderate recurrent hypoglycemia. Diabetes 55:1088–1095
Figlewicz DP, Van Dijk G, Wilkinson CW, Gronbeck P, Higgins M, Zavosh A (2002) Effects of repetitive hypoglycemia on neuroendocrine response and brain tyrosine hydroxylase activity in the rat. Stress 5:217–226
Stanciu M, Radulovic J, Spiess J (2001) Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Brain Res Mol Brain Res 94:15–24
Alberini CM (2009) Transcription factors in long-term memory and synaptic plasticity. Physiol Rev 89:121–145
Martin S, Henley JM, Holman D et al (2009) Corticosterone alters AMPAR mobility and facilitates bidirectional synaptic plasticity. PLoS One 4, e4714
Tse YC, Bagot RC, Wong TP (2012) Dynamic regulation of NMDAR function in the adult brain by the stress hormone corticosterone. Front Cell Neurosci 6:9
Barco A, Alarcon JM, Kandel ER (2002) Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108:689–703
Guzowski JF, Setlow B, Wagner EK, McGaugh JL (2001) Experience-dependent gene expression in the rat hippocampus after spatial learning: a comparison of the immediate-early genes Arc, c-fos, and zif268. J Neurosci : Off J Soc Neurosci 21:5089–5098
Davis SN (2009) Diabetes: Hypoglycemia–a new approach to an old problem. Nat Rev Endocrinol 5:243–245
Acknowledgements
We thank L. Jacobson (Professor, Albany Medical College, Albany, NY) and A. Poulos (Assistant Professor, University at Albany, Albany, NY) for insightful comments on the analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding for this work came from the American Diabetes Association (7-12-BS-126 to E. C. McNay), NIA (R01 AG050598 to E. C. McNay) and NIDDK (R01 077106 to E. C. McNay).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contributions
All authors made substantial contributions to the design, acquisition of data, analysis and interpretation of data, as well as helping in the drafting and revision of this manuscript. All authors have given final approval of this version to be published. ECM is the guarantor of this work as a whole.
Rights and permissions
About this article

Cite this article
Osborne, D.M., O’Leary, K.E., Fitzgerald, D.P. et al. Context-dependent memory following recurrent hypoglycaemia in non-diabetic rats is mediated via glucocorticoid signalling in the dorsal hippocampus. Diabetologia 60, 182–191 (2017). https://doi.org/10.1007/s00125-016-4114-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4114-1