
Abstract
Diabetic kidney disease (DKD) is the most common cause of kidney failure in many countries today, but treatments have not improved in the last 20 years. Recently, systems biology methods have allowed the elucidation of signalling pathways and networks involved in the progression of DKD that were not well appreciated previously. A prominent pathway found to be integrally associated with DKD progression is the Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathway. Increased expression of JAK–STAT genes was found in multiple cells in the kidney, including glomerular podocytes, in both early and progressive DKD. Subsequent experiments in a mouse diabetic model showed that enhanced expression of JAK2 selectively in glomerular podocytes increased functional and pathological features of DKD. Finally, a yet unpublished Phase 2 multicentre, randomised, double-blind, placebo-controlled study of the efficacy of a selective JAK1 and JAK2 inhibitor has been conducted in type 2 diabetic participants with DKD. In this trial there was a reduction of albuminuria in participants who received the active inhibitor compared with those who received a placebo These results support the further study of JAK inhibitors as a new therapy for DKD. This review summarises a presentation given at the ‘Anti-inflammatory interventions in diabetes’ symposium at the 2015 annual meeting of the EASD. It is accompanied by an overview by the Session Chair, Hiddo Heerspink (DOI: 10.1007/s00125-016-4030-4).
Similar content being viewed by others
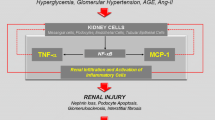


Diabetic kidney disease (DKD) is the most common cause of end-stage renal disease (ESRD) in the USA and in many other countries today [1]. Furthermore, its incidence continues to increase because of the epidemic of type 2 diabetes, despite some recent improvement in the rates of progressive DKD [1]. In spite of the unquestionable salutary impact of blood pressure control and pharmacological inhibition of the renin–angiotensin–aldosterone system (RAAS), additional powerful interventions are needed in order to allow us to make any real headway in the fight against the progression of DKD. To address this challenge, our team has analysed transcriptomic profiles of kidney samples from humans with progressive DKD and those of murine DKD models to identify new pathways involved in the progression of DKD, to uncover potential new targets for intervention that might add to the effects of RAAS inhibition.
In these studies we found that the expression of multiple Janus kinase–signal transducer and activator of transcription (JAK–STAT) family members was substantially increased in kidney tissues from humans with DKD [2] (Table 1). JAK–STAT proteins transduce signals from several types of receptor. After their ligands bind to them, these membrane receptors dimerise, recruit pairs of JAK proteins to the intracellular receptor domains and activate them via autophosphorylation. Activated JAKs then phosphorylate STAT proteins which form hetero- or homodimers and translocate to the nucleus to induce the transcription of target genes [3].
We found that multiple JAK and STAT isoforms, including JAK2, were expressed at higher levels in glomeruli and tubulointerstitial regions from patients with DKD than in samples from controls [2] (Table 1). The pattern of increased expression was quite informative. In patients with early DKD and normal kidney function, there was intense glomerular overexpression of the various JAK and STAT isoforms. However, in patients with progressive DKD with declining kidney function, glomerular JAK and STAT mRNA expression mostly returned towards normal while tubulointerstitial expression increased substantially. Moreover, the tubulointerstitial expression of each of the JAK and STAT isoforms was significantly inversely correlated with the estimated GFR in these patients with DKD. This temporal difference in kidney regional expression of the JAK–STAT mRNAs corresponded to the DKD disease process, whereby pathology begins in the glomerulus and then progresses to the tubulointerstitium [2]. We also found that expression of JAK2 protein in proximal tubule cells was high in patients with progressive kidney disease compared with that in patients without kidney disease and even compared with that in a few individuals with other glomerular diseases. JAK2 protein was also expressed in glomerular cells, including podocytes. This pattern of expression suggested that the changes in JAK2 expression in DKD reflected changes in intrinsic renal cells rather than in infiltrating inflammatory cells [2].
In a separate study, we found transcriptomic evidence for generalised JAK–STAT activation, but not increases in JAK–STAT gene expression, in glomeruli from three mouse models of DKD in cross-species network analyses [4]. This pattern was shared in glomerular samples from patients with type 2 diabetes and very early kidney disease [4]. The JAK–STAT pathway was the most prominent of the pathways in terms of numbers of genes with increased expression among humans and all three mouse models.
To determine whether the enhanced JAK–STAT expression seen in glomerular podocytes in early human DKD contributes to the pathology and early progression of diabetic glomerulopathy, we overexpressed JAK2 specifically in podocytes of mice in the presence and absence of diabetes. We found that overexpression of podocyte JAK2 had little effect in non-diabetic mice, but in diabetic mice it resulted in markedly increased glomerular functional and pathological features of DKD including albuminuria, mesangial expansion, reduction in podocyte density, glomerulosclerosis and glomerular basement membrane thickening . These features were improved dramatically by 2 weeks of treatment with an oral JAK1 and JAK2 inhibitor [5].
Other investigators have confirmed the enhanced activity of the JAK–STAT pathway in animal models of DKD. For example, transgenic mice with reduced capacity for STAT3 activation developed less proteinuria, mesangial expansion, cell proliferation, macrophage infiltration, inflammation and abnormal matrix synthesis when made diabetic by the administration of streptozotocin [6]. Other important regulators of JAK–STAT signalling are the suppressors of cytokine signalling (SOCS). SOCS1 and SOCS3 are downstream transcriptional targets of the JAK–STAT signalling pathway that bind to JAK proteins and interfere with their activity in a negative-feedback manner [7]. Rats injected with recombinant SOCS1 and SOCS3 adenovirus showed evidence of reduced JAK/STAT activation and amelioration of early diabetic changes after 7 weeks of diabetes [8]. Finally, another study reported that STAT3 acetylation is increased in both mouse and human diabetic kidneys [9]. This has functional implications because acetylation of STAT proteins enhances STAT dimerisation, which is critical for the translocation of STATs to the nucleus and their ability to modulate gene transcription [10]. Treatment with inhibitors that block acetylation-mediated association of STAT3 reduced both proteinuria and kidney injury in the db/db mouse model of diabetes.
Based on the growing evidence for JAK–STAT activation in DKD, baricitinib, a selective JAK1 and JAK2 inhibitor, developed by Eli Lilly and Company and Incyte for other indications, was investigated in a Phase 2 multicentre, randomised, double-blind, multi-dose, placebo-controlled study (NCT01683409) in participants with type 2 diabetic nephropathy with reduced kidney function (eGFR 25–75 ml min–1 1.73 m−2) and substantial persistent albuminuria (>300 mg/day) who were already treated with RAAS-inhibiting agents. Participants received placebo or barcitinib at low-to-high daily doses (0.75 mg, 1.5 mg in single or divided dose, 4 mg) for 24 weeks, followed by a 4-week wash-out period. The primary efficacy measure for this trial was the change in urine albumin to creatinine ratio (UACR) from baseline to 6 months of treatment. Secondary measures included UACR change from baseline to 3 months, 24 h albumin and total protein excretion at 6 months, change in plasma cystatin C levels from baseline to 6 months, biomarkers and safety indicators. Interestingly, despite the fact that multiple centres in both Japan and North America were involved in the study, it was difficult to identify eligible participants; this was largely due to the fact that most North American candidates who were screened failed to meet the albuminuria threshold of >300 mg/day. Planned enrolment was 250 participants. However, we terminated enrolment when a blinded sample size reassessment indicated that statistical significance could be achieved with a smaller number of patients. The study was completed with 129 participants randomised and treated for 24 weeks.
Baricitinib treatment resulted in a reduction in albuminuria at both 3 and 6 months. After 4 weeks of study drug wash-out, the albuminuria reduction was sustained in the medium- and high-dose baricitinib groups. The magnitude of this reduction was approximately 40% vs placebo in the highest dose group. Importantly, there was evidence of target engagement and a reduction in renal inflammation with baricitinib treatment, as assessed by urinary levels of IFNγ-induced protein 10 (IP-10) and plasma levels of TNF receptor 2 (TNFR2) [11]. The changes in TNFR2 are especially meaningful, as elevated plasma TNFR2 and TNFR1 levels have been shown to be biomarkers with high prognostic power for progression of DKD in both type 1 and type 2 diabetic populations [12, 13]. No change in kidney function at 6 months occurred with baricitinib treatment based on estimated GFR calculated using serum cystatin C levels. At 6 months, there was a statistically significant decrease in haemoglobin compared with placebo in the high-dose baricitinib group. This reduction was not unexpected in the higher dose range since erythropoietin signalling is dependent on JAK2 activation [14]. No unexpected side effects were detected. These data were presented in preliminary form at the American Diabetes Association meeting in June 2015 [11]. Further studies will need to examine the effects of baricitinib on progression of DKD as determined by harder endpoints, such as eGFR decline, ESRD and mortality events.
The growing evidence for JAK–STAT activation in the pathogenesis of DKD establishes a new set of targets for potential intervention in this disease. Whether baricitinib or other JAK inhibitors will show a positive effect on the progression of DKD in terms of loss of kidney function and development of ESRD and other complications remains uncertain, though the preclinical and early clinical data are quite encouraging. Another question is whether long-term administration of JAK inhibitors in the treatment of DKD will be safe. These agents were developed for the treatment of autoimmune conditions as well as myelodysplastic syndrome, and treatment to date with any of the inhibitors has been of significantly shorter duration than the many years of treatment that effective DKD therapy would be likely to entail. The long-term effect of JAK2 inhibition may be problematic if the treatment potentially aggravates the anaemia that many DKD patients have. Finally, as noted earlier in this review, diabetes can result in activation of STAT proteins independently of JAK activation, suggesting that at least some of the STAT-dependent processes would be independent of JAK inhibition and that intervention more distally in the signalling cascade may also be necessary. Despite these concerns, there is good reason to be optimistic about the outcomes of long-term JAK inhibition as a treatment that could substantially slow the progression of DKD and make outcomes better for the millions of individuals worldwide who suffer from this often fatal disease.
Abbreviations
- DKD:
-
Diabetic kidney disease
- ESRD:
-
End-stage renal disease
- JAK:
-
Janus kinase
- RAAS:
-
Renin–angiotensin–aldosterone system
- SOCS:
-
Suppressors of cytokine signalling
- STAT:
-
Signal transducer and activator of transcription
- TNFR:
-
TNF receptor
- UACR:
-
Urine albumin to creatinine ratio
References
Saran R, Li Y, Robinson B et al (2015) US Renal Data System 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis 66(1 Suppl 1):Svii, S1-S305
Berthier CC, Zhang H, Schin M et al (2009) Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 58:469–477
Brosius FC 3rd, He JC (2015) JAK inhibition and progressive kidney disease. Curr Opin Nephrol Hypertens 24:88–95
Hodgin JB, Nair V, Zhang H et al (2013) Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 62:299–308
Zhang H, Saha J, Atkins KB, Brosius FC (2012) Podocyte JAK2 augments glomerular injury induced by diabetes and angiotensin II. J Am Soc Nephrol: 23:203A (TH-PO461) (Abstract)
Lu TC, Wang ZH, Feng X et al (2009) Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int 76:63–71
Croker BA, Kiu H, Nicholson SE (2008) SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol 19:414–422
Õrtiz-Munoz G, Lopez-Parra V, Lopez-Franco O et al (2010) Suppressors of cytokine signaling abrogate diabetic nephropathy. J Am Soc Nephrol 21:763–772
Liu R, Zhong Y, Li X et al (2014) Role of transcription factor acetylation in diabetic kidney disease. Diabetes 63:2440–2453
Zhuang S (2013) Regulation of STAT signaling by acetylation. Cell Signal 25:1924–1931
Tuttle K, Brosius F, Adler S et al (2015) Baricitinib in diabetic kidney disease: results from a phase 2, multi-center, randomized, double-blind, placebo-controlled study. Diabetes 64(Suppl 1): Abstract no. 114-LB. Available from http://app.core-apps.com/tristar_ada15/abstract/1678f1629f1672a1656ce1635baedcc1625f3994ccb, accessed 20 May 2016
Niewczas MA, Gohda T, Skupien J et al (2012) Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol 23:507–515
Gohda T, Niewczas MA, Ficociello LH et al (2012) Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol 23:516–524
Kuhrt D, Wojchowski DM (2015) Emerging EPO and EPO receptor regulators and signal transducers. Blood 125:3536–3541
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
These studies were supported in part by grants from the National Institutes of Health (NIDDK R24DK082841, George O’Brien Kidney Core Center, NIDDK P30 DK081943), and the Juvenile Diabetes Research Foundation (JDRF, 2-SRA-2014-257-Q-R).
Duality of interest
The Phase 2 clinical trial of the efficacy of baricitinib in the treatment of diabetic kidney disease was funded by Eli Lilly and Co. All three authors were on the Steering Committee of this study. KRT and FCB were Principal Investigators of the study.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Rights and permissions
About this article

Cite this article
Brosius, F.C., Tuttle, K.R. & Kretzler, M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia 59, 1624–1627 (2016). https://doi.org/10.1007/s00125-016-4021-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4021-5