
Abstract
In recent years, novel discoveries have reshaped our understanding of the biology of brain glucagon in the regulation of peripheral homeostasis. Here we compare and contrast brain glucagon action in feeding vs glucose regulation and depict the physiological relevance of brain glucagon by reviewing their actions in two key regions of the central nervous system: the mediobasal hypothalamus and the dorsal vagal complex. These novel findings pave the way to future therapeutic strategies aimed at enhancing brain glucagon action for the treatment of diabetes and obesity. This review summarises a presentation given at the ‘Novel data on glucagon’ symposium at the 2015 annual meeting of the EASD. It is accompanied by two other reviews on topics from this symposium (by Young Lee and colleagues, DOI: 10.1007/s00125-016-3965-9), and by Russell Miller and Morris Birnbaum, DOI: 10.1007/s00125-016-3955-y) and an overview by the Session Chair, Isabel Valverde (DOI: 10.1007/s00125-016-3946-z).
Similar content being viewed by others


Introduction
In recent decades, the notion of a central nervous system-mediated pathway for the physiology of glucagon has been garnering scientific attention. Multiple regions in the brain have demonstrated significant binding to radiolabelled glucagon via glucagon receptors as well as transduced activation of adenylate cyclase [1]. Indeed, glucagon is capable of permeating the blood–brain barrier [2], while significant glucagon immunoreactivity has also been detected mostly in the hypothalamus and brainstem regions, two sites known to modulate peripheral metabolism [3]. Furthermore, the classical components of the glucagon receptor signalling cascade, including cAMP and protein kinase A (PKA) are also detected in brain neurons [4], thus suggesting the possibility that glucagon could trigger its signalling cascade in the brain to regulate peripheral homeostasis.
In fact, multiple studies have documented the ability of intracerebroventricular (i.c.v.) glucagon administration to modulate peripheral glucose levels in different species of animals [5–7]. Most intriguing, however, is the earliest study in dogs where a high dose i.c.v. injection of 10 ng of glucagon transiently produced hypoglycaemia followed by hyperglycaemia [8]. The hypoglycaemic effect was abolished in vagotomised dogs, suggesting the involvement of a brain–liver axis in the glucose-lowering effect of central glucagon, whereas pancreatectomy prevented the hyperglycaemic effect, attributing a pancreatic role to the rise of glucose from i.c.v. glucagon injections [8, 9]. Given the non-specific administration of i.c.v. glucagon and the use of relatively high glucagon dose in these experiments, more recent studies have administered much lower doses of glucagon specifically into the mediobasal hypothalamus (MBH) and evaluated whether MBH glucagon action accounts for the glucose-lowering effect or hyperglycaemic effect of i.c.v. injections in the early dog studies.
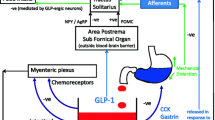
Indeed, direct infusion of glucagon into the MBH actually reduced hepatic glucose production under conditions of a pancreatic basal insulin euglycaemic clamp [10]. This effect of MBH glucagon required the activation of MBH glucagon receptors (GRs), PKA signalling, KATP channels (Fig. 1) and intact vagal nerves, since their ablation negated the metabolic effects of MBH glucagon infusion [10, 11]. On the other hand, blocking lipid sensing-dependent pathways in MBH at the level of 5′ AMP-activated protein kinase (AMPK) or protein kinase C-δ did not reverse the effect of MBH glucagon, suggesting that MBH glucagon regulates peripheral glucose metabolism via a lipid sensing-independent mechanism (Fig. 1).

Glucagon action in the MBH and the DVC regulates glucose and energy homeostasis. Schematic representation illustrating that glucagon action in the MBH triggers differential signalling pathways to exert effects on feeding (AMPK-dependent) and glucose (AMPK-independent) control. While it remains unknown whether glucagon action in the DVC regulates feeding, glucagon initiates a GR–PKA–ERK1/2–KATP signalling cascade to lower hepatic glucose production. The brain also mediates the effect of elevated circulating glucagon induced either by i.v. glucagon or high-protein meals to maintain glucose homeostasis in rodents in vivo. CPT-1, carnitine palmitoyltransferase I; ERK1/2, extracellular signal-regulated kinase 1/2; LCFA, long-chain fatty acid
Multiple groups have also confirmed a similar paradigm of i.c.v. glucagon action in feeding behaviour regulation. In studies in rats, chicks and sheep, i.c.v. administration of glucagon reduced food intake [12–14]. Although far less is known about the mechanisms underlying these anorectic effects of brain glucagon, an interesting recent study revealed novel mechanisms by which hypothalamic glucagon action impacts feeding [15]. This invited review will focus on comparing and contrasting brain glucagon action in feeding vs glucose control (Fig. 1).
Hypothalamic glucagon action in feeding vs glucose control
Experiments conducted by Quiñones and colleagues demonstrated that central glucagon injection in rats led to significantly reduced feeding behaviour through the action of glucagon in the hypothalamus [15]. Similar to its effect on glucose homeostasis [10], hypothalamic glucagon requires GRs and activation of downstream PKA in the hypothalamic arcuate nucleus (ARC) to carry out its anorectic effects (Fig. 1), since i.c.v. co-infusion of the GR antagonist des-His1-[Glu9] glucagon amide or the PKA inhibitor H-89 negated the ability of central glucagon to decrease feeding. Of note, these were the same chemical inhibitors employed to confirm the role of GR and PKA in mediating MBH glucagon action on glucose control [10], further suggesting that any associated molecular players in the GR–PKA branch are likely to be a part of a common pathway for both feeding and glucose regulation by brain glucagon. Moreover, Quiñones et al [15] also reported changes in agouti-related peptide (AgRP) expression associated with the anorectic action of central glucagon, again, consistent with the observation that MBH GRs co-localised with AgRP neurons [10]. These findings point to the idea that, unlike in brain insulin action, where AgRP neurons are necessary for glucose, but not energy, homeostasis [16], in brain glucagon action, AgRP neurons mediate both glucose and feeding regulation (Fig. 1).
However, it appears that not the entire signalling pathway converges for the glycaemic and satiety effects of hypothalamic glucagon action. Contrary to how MBH glucagon signals to exert glucose control independent of MBH AMPK [11], the suppressive effect central glucagon exerts on feeding involved inhibition of AMPK and activation of downstream target acetyl-CoA carboxylase (ACC) [15]. Specifically, molecular activation of AMPK in the ARC via injection of a constitutively active AMPK virus blunted the anorectic effects of central glucagon injections [15], whereas activation of MBH AMPK via the same viral approach had no effect on the glucose production-lowering effect of MBH glucagon [11]. Consistent with this, there were decreased AMPK and increased ACC in the ARC associated with the satiety effect of central glucagon injections [15], whereas no differences in pACC/total ACC levels in the MBH were associated with the glucose-lowering effect of MBH glucagon infusions [11]. While these findings illustrate the distinct mechanisms by which brain glucagon acts, it cannot be overlooked that perhaps the differences in glucagon dose and administration—a single bolus i.c.v. glucagon injection at a dose of 480 ng for feeding [15] vs 2 h of constant MBH glucagon infusion with a much lower dose of 3.6 pg for glucose control [11]—could explain some of the differences in the regulation of molecular targets for feeding and glucose regulation by brain glucagon. Alternatively, these findings could be due to differences in the times at which the glucagon-treated tissues were obtained for molecular analysis: changes in AMPK and ACC, which mediate the anorectic action of hypothalamic glucagon, were measured 1 h after the i.c.v. glucagon single bolus injection vs after 2 h of constant infusion of MBH glucagon.
Interestingly, one study has reported that a 10 min i.v. infusion of glucagon inhibits food intake in humans [17], suggesting that the effects of glucagon action in the hypothalamus of rodents to lower food intake, as discussed above, may have mediated the anorectic effect of i.v. glucagon infusion in humans. With respect to glucose control, a continuous i.v. glucagon infusion rapidly increases blood glucose levels and glucose production in rats, dogs and humans [10, 18–20]. However, this effect of glucagon on glucose regulation is transient in nature, with both glucose levels and glucose production declining to baseline within 40 min, despite the continuing hyperglucagonaemia achieved by i.v. glucagon infusion. Importantly, direct blockade of hypothalamic glucagon signalling abolishes the transient effect of i.v. glucagon infusion on glucose production in rats, indicating that hypothalamic glucagon action is physiologically responsible for countering the direct hepatic stimulatory effect on glucose production by glucagon [10] (Fig. 1). Although the physiological relevance of hypothalamic glucagon signalling in mediating the effect of circulating glucagon to regulate food intake and glucose homeostasis warrants further investigation, mounting evidence also suggests that hypothalamic glucagon signalling is pathologically relevant, at least in rodents.
High-fat feeding induces hypothalamic glucagon resistance in both glucose and feeding controls [10, 15]. The ability of MBH glucagon infusion to lower glucose production was abolished in rats fed a high-fat diet for 3 days [10]. Similarly, high-fat feeding for 3 weeks also blocked the hypophagic effect induced by glucagon signalling [15]. While chemical activation of PKA using Sp-cyclic adenosine-3′, 5′-monophosphorothioate (Sp-cAMPS) in the MBH was effective in restoring the glucose production-lowering action of MBH glucagon in the 3 day high-fat fed model [10], Sp-cAMPS failed to restore glucagon sensitivity in the ARC to inhibit feeding in the rats fed a high-fat diet for 3 weeks. On the other hand, it was restored by molecular inhibition of hypothalamic Ca2+-calmodulin-dependent protein kinase kinase β (CaMKKβ) via adenoviruses encoding dominant negative CaMKKβ [15]. Collectively, these findings suggest that, in the context of feeding control, hypothalamic glucagon resistance is manifested by the inability of PKA to inhibit CaMKKβ, whereas in glucose control, the site of the defect lies upstream of PKA activation (Fig. 1). Alternatively, the difference in the site of the defect with respect to brain glucagon action could also be due to differences in the duration of high-fat feeding [10, 15].
Dorsal vagal complex glucagon action in glucose control
Following these novel findings of effects on peripheral glucose levels and energy balance mediated by glucagon action within the hypothalamus, it became important to question whether glucagon can also act in other sites of the brain to influence peripheral homeostasis. The dorsal vagal complex (DVC), located in the brainstem, is another key brain site known to regulate several autonomic functions, including peripheral glucose and energy homeostasis, via neural and hormonal signals [21]. Indeed, studies have confirmed that insulin [22, 23], glucagon-like peptide 1 (GLP-1) [24, 25] and leptin [26] signalling in the DVC regulate glycaemia and/or feeding behaviour. In light of the fact that glucagon receptors are also abundantly expressed in the brainstem [1, 27], and peripherally administered glucagon alters neuronal activity in the brainstem [28], it was plausible that, analogous to the MBH, glucagon could initiate a signalling cascade in the DVC to regulate peripheral metabolism.
While it remains to be investigated whether glucagon acts in the DVC to regulate food intake (Fig. 1), a recent study has revealed that glucagon action in the DVC can impact glucose homeostasis, and has postulated a novel physiological role of DVC glucagon action in postprandial conditions [29]. During pancreatic basal insulin euglycaemic clamp conditions, glucagon infusion into the DVC reduced hepatic glucose production in healthy rodents [29]. Using complementary chemical and molecular loss-of-function approaches, blocking activation of the GR, PKA, extracellular signal-regulated kinase (ERK) 1/2 or KATP channels in the DVC reversed the glucose production-lowering effect of DVC glucagon infusion, thereby implicating each of these as the downstream signalling mediators in the DVC glucagon signalling pathway (Fig. 1). The physiological relevance of DVC glucagon action in regulating glucose homeostasis was investigated in the context of the postprandial period following high-protein meals [29].
High-protein feeding acutely lowers the postprandial glucose response, despite significant stimulation of circulating glucagon levels [30–33]. This apparent discrepancy, given that peripheral glucagon classically increases glycaemia, could be explained by an additional mechanism counter the effect of hepatic glucagon action during high-protein feeding so as to maintain glucose homeostasis. The fact that the DVC is accessible to peripheral nutrients and hormones [34, 35] suggested that following high-protein consumption, elevated glucagon could reach the DVC to activate its GR-mediated signalling pathway to lower hepatic glucose production and thereby lower plasma glucose levels.
In fact, during fasting–refeeding experiments, high-protein feeding acutely decreased plasma glucose levels compared with a low-protein diet; but when DVC GR signalling was blocked, it negated the ability of high- vs low-protein feeding to suppress the rise in plasma glucose [29]. Thus, indeed, it is the action of the elevated circulating glucagon in the DVC that lowers plasma glucose during high-protein feeding (Fig. 1). Perhaps then, could the action of circulating glucagon in the DVC also be responsible for the satiety effect of high-protein meals previously observed in humans and rodents [36, 37] (Fig. 1)?
Conclusion and future perspective
In summary, the brain is increasingly being recognised as an important glucagon-sensitive organ. In contrast to the hormone’s systemic effect on glucose response, glucagon action in the MBH and DVC actually reduces hepatic glucose production. These central sites are also sensitive to circulating glucagon during conditions such as i.v. glucagon infusion and high-protein feeding, which serves to antagonise the hepatic actions of the hormone to maintain glucose response (Fig. 1). Furthermore, the role of hypothalamic glucagon is also implicated in reducing food intake, via common as well as distinct signalling mechanisms that mediate glucose control (Fig. 1).
These novel findings indicate that maintaining glucagon action sufficient to activate the central glucagon system may be therapeutically advantageous. In fact, multiple studies have already reported that activation of glucagon receptors in conjunction with other G protein-coupled receptors is metabolically advantageous in diabetes and obesity. For example, a triagonist aimed at simultaneous activation of glucagon, GLP-1 and glucose-dependent insulinotropic (GIP) receptors improved metabolic and glycaemic profiles in obese and diabetic rodents [38]. In addition, work on dual activation of glucagon and GLP-1 receptors normalised glucose tolerance and reduced food intake in mice with diet-induced obesity [39, 40]. While these pre-clinical studies have attributed the beneficial effects of glucagon primarily to increased energy expenditure and decreased food intake and its hyperglycaemic effects to be countered by the actions of GLP-1 and/or GIP, the brain penetrance of these polyagonists and whether activation of central glucagon signalling plays a role in counteracting the diabetogenic effects of peripheral glucagon in these polyagonist therapeutic strategies remain to be investigated.
Abbreviations
- ACC:
-
Acetyl CoA carboxylase
- AgRP:
-
Agouti-related peptide
- ARC:
-
Arcuate nucleus
- CaMKKβ:
-
Ca2+-calmodulin-dependent protein kinase kinase β
- DVC:
-
Dorsal vagal complex
- GLP-1:
-
Glucagon-like peptide 1
- GR:
-
Glucagon receptor
- i.c.v.:
-
Intracerebroventricular
- MBH:
-
Mediobasal hypothalamus
- PKA:
-
Protein kinase A
- Sp-cAMPS:
-
Sp-cyclic adenosine-3′, 5′-monophosphorothioate
References
Hoosein NM, Gurd RS (1984) Identification of glucagon receptors in rat brain. Proc Natl Acad Sci U S A 81:4368–4372
Banks WA, Kastin AJ (1985) Peptides and the blood-brain barrier: lipophilicity as a predictor of permeability. Brain Res Bull 15:287–292
Sasaki H, Ebitani I, Tominaga M, Yamatani K, Yawata Y, Hara M (1980) Glucagon-like substance in the canine brain. Endocrinol Jpn 27(Suppl 1):135–140
Wetsel WC, Eraly SA, Whyte DB, Mellon PL (1993) Regulation of gonadotropin-releasing hormone by protein kinase-A and -C in immortalized hypothalamic neurons. Endocrinology 132:2360–2370
Marubashi S, Tominaga M, Katagiri T et al (1985) Hyperglycaemic effect of glucagon administered intracerebroventricularly in the rat. Acta Endocrinol (Copenh) 108:6–10
Honda K, Kamisoyama H, Uemura T et al (2012) The mechanism underlying the central glucagon-induced hyperglycemia and anorexia in chicks. Comp Biochem Physiol A Mol Integr Physiol 163:260–264
Amir S (1986) Central glucagon-induced hyperglycemia is mediated by combined activation of the adrenal medulla and sympathetic nerve endings. Physiol Behav 37:563–566
Agarwala GC, Bapat SK (1977) Effect of centrally administered glucagon on blood glucose levels in dogs. Indian J Med Res 66:323–330
Agarwala GC, Mishra R, Jaiswal G, Bapat V (1989) Effect of centrally administered glucagon on liver glycogen & enzymes in anaesthetised dogs. Indian J Med Res 90:372–378
Mighiu PI, Yue JT, Filippi BM et al (2013) Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat Med 19:766–772
Abraham MA, Yue JT, LaPierre MP et al (2014) Hypothalamic glucagon signals through the KATP channels to regulate glucose production. Mol Metab 3:202–208
Inokuchi A, Oomura Y, Nishimura H (1984) Effect of intracerebroventricularly infused glucagon on feeding behavior. Physiol Behav 33:397–400
Honda K, Kamisoyama H, Saito N, Kurose Y, Sugahara K, Hasegawa S (2007) Central administration of glucagon suppresses food intake in chicks. Neurosci Lett 416:198–201
Kurose Y, Kamisoyama H, Honda K et al (2009) Effects of central administration of glucagon on feed intake and endocrine responses in sheep. Anim Sci J 80:686–690
Quiñones M, Al-Massadi O, Gallego R et al (2015) Hypothalamic CaMKKβ mediates glucagon anorectic effect and its diet-induced resistance. Mol Metab 4:961–970
Könner AC, Janoschek R, Plum L et al (2007) Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 5:438–449
Geary N, Kissileff HR, Pi-Sunyer FX, Hinton V (1992) Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men. Am J Physiol 262:R975–R980
Bomboy JD Jr, Lewis SB, Lacy WW, Sinclair-Smith BC, Liljenquist JE (1977) Transient stimulatory effect of sustained hyperglucagonemia on splanchnic glucose production in normal and diabetic man. Diabetes 26:177–184
Felig P, Wahren J, Hendler R (1976) Influence of physiologic hyperglucagonemia on basal and insulin-inhibited splanchnic glucose output in normal man. J Clin Invest 58:761–765
Eigler N, Sacca L, Sherwin RS (1979) Synergistic interactions of physiologic increments of glucagon, epinephrine, and cortisol in the dog: a model for stress-induced hyperglycemia. J Clin Invest 63:114–123
Abraham MA, Filippi BM, Kang GM, Kim MS, Lam TK (2014) Insulin action in the hypothalamus and dorsal vagal complex. Exp Physiol 99:1104–1109
Filippi BM, Yang CS, Tang C, Lam TK (2012) Insulin activates Erk1/2 signaling in the dorsal vagal complex to inhibit glucose production. Cell Metab 16:500–510
Filippi BM, Bassiri A, Abraham MA, Duca FA, Yue JT, Lam TK (2014) Insulin signals through the dorsal vagal complex to regulate energy balance. Diabetes 63:892–899
Hayes MR, Skibicka KP, Grill HJ (2008) Caudal brainstem processing is sufficient for behavioral, sympathetic, and parasympathetic responses driven by peripheral and hindbrain glucagon-like-peptide-1 receptor stimulation. Endocrinology 149:4059–4068
Hayes MR, Bradley L, Grill HJ (2009) Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. Endocrinology 150:2654–2659
Hayes MR, Skibicka KP, Leichner TM et al (2010) Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab 11:77–83
Hansen LH, Abrahamsen N, Nishimura E (1995) Glucagon receptor mRNA distribution in rat tissues. Peptides 16:1163–1166
Parker JA, McCullough KA, Field BC et al (2013) Glucagon and GLP-1 inhibit food intake and increase c-fos expression in similar appetite regulating centres in the brainstem and amygdala. Int J Obes (Lond) 37:1391–1398
LaPierre MP, Abraham MA, Yue JT, Filippi BM, Lam TK (2015) Glucagon signalling in the dorsal vagal complex is sufficient and necessary for high-protein feeding to regulate glucose homeostasis in vivo. EMBO Rep 16:1299–1307
Calbet JA, MacLean DA (2002) Plasma glucagon and insulin responses depend on the rate of appearance of amino acids after ingestion of different protein solutions in humans. J Nutr 132:2174–2182
Claessens M, Calame W, Siemensma AD, van Baak MA, Saris WH (2009) The effect of different protein hydrolysate/carbohydrate mixtures on postprandial glucagon and insulin responses in healthy subjects. Eur J Clin Nutr 63:48–56
Day JL, Johansen K, Ganda OP, Soeldner JS, Gleason RE, Midgley W (1978) Factors governing insulin and glucagon responses during normal meals. Clin Endocrinol (Oxf) 9:443–454
Peret J, Foustock S, Chanez M, Bois-Joyeux B, Assan R (1981) Plasma glucagon and insulin concentrations and hepatic phosphoenolpyruvate carboxykinase and pyruvate kinase activities during and upon adaptation of rats to a high protein diet. J Nutr 111:1173–1184
Cheung GW, Kokorovic A, Lam CK, Chari M, Lam TK (2009) Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metab 10:99–109
Wang PY, Caspi L, Lam CK et al (2008) Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature 452:1012–1016
Weigle DS, Breen PA, Matthys CC et al (2005) A high-protein diet induces sustained reductions in appetite, ad libitum caloric intake, and body weight despite compensatory changes in diurnal plasma leptin and ghrelin concentrations. Am J Clin Nutr 82:41–48
Blouet C, Mariotti F, Azzout-Marniche D et al (2006) The reduced energy intake of rats fed a high-protein low-carbohydrate diet explains the lower fat deposition, but macronutrient substitution accounts for the improved glycemic control. J Nutr 136:1849–1854
Finan B, Yang B, Ottaway N et al (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21:27–36
Pocai A, Carrington PE, Adams JR et al (2009) Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes 58:2258–2266
Day JW, Ottaway N, Patterson JT et al (2009) A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol 5:749–757
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The work from the TKTL laboratory discussed in this mini-review is funded by a Canadian Diabetes Association Operating Grant (OG-3-15-4901-TL). MAA is supported by a Canadian Institutes of Health Research Doctoral Award. TKTL holds the John Kitson McIvor (1915–1942) Endowed Chair in Diabetes Research and Canada Research Chair in Obesity at the Toronto General Research Institute and the University of Toronto.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
TKTL and MAA drafted the article, revised it critically for important intellectual content and approved the version to be published.
Rights and permissions
About this article

Cite this article
Abraham, M.A., Lam, T.K.T. Glucagon action in the brain. Diabetologia 59, 1367–1371 (2016). https://doi.org/10.1007/s00125-016-3950-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3950-3