
Abstract
Aims/hypothesis
The crosstalk between skeletal muscle (SkM) and beta cells plays a role in diabetes aetiology. In this study, we have investigated whether SkM-released exosome-like vesicles (ELVs) can be taken up by pancreatic beta cells and can deliver functional cargoes.
Methods
Mice were fed for 16 weeks with standard chow diet (SCD) or with standard diet enriched with 20% palmitate (HPD) and ELVs were purified from quadriceps muscle. Fluorescent ELVs from HPD or SCD quadriceps were injected i.v. or intramuscularly (i.m.) into mice to determine their biodistributions. Micro (mi)RNA quantification in ELVs was determined using quantitative real-time RT-PCR (qRT-PCR)-based TaqMan low-density arrays. Microarray analyses were performed to determine whether standard diet ELVs (SD-ELVs) and high palmitate diet ELVs (HPD-ELVs) induced specific transcriptional signatures in MIN6B1 cells.
Results
In vivo, muscle ELVs were taken up by pancreas, 24 h post-injection. In vitro, both SD-ELVs and HPD-ELVs transferred proteins and miRNAs to MIN6B1 cells and modulated gene expressions whereas only HPD-ELVs induced proliferation of MIN6B1 cells and isolated islets. Bioinformatic analyses suggested that transferred HPD-ELV miRNAs may participate in these effects. To validate this, we demonstrated that miR-16, which is overexpressed in HPD-ELVs, was transferred to MIN6B1 cells and regulated Ptch1, involved in pancreas development. In vivo, islets from HPD mice showed increased size and altered expression of genes involved in development, including Ptch1, suggesting that the effect of palm oil on islet size in vivo was reproduced in vitro by treating beta cells with HPD-ELVs.
Conclusions/interpretation
Our data suggest that muscle ELVs might have an endocrine effect and could participate in adaptations in beta cell mass during insulin resistance.
Similar content being viewed by others
Introduction
Long distance transmission of signals between tissues is commonly believed to be associated with protein-based signalling systems, such as enzymes, hormones, cytokines and chemokines. In this context, skeletal muscle (SkM)-secreted proteins play an important role in intercellular communications [1, 2]. Recently, it has been demonstrated that muscle cells also release membranous bioactive extracellular vesicles (EVs) (exosomes, macroparticles and apoptotic bodies), which transfer both functional proteins and RNA species between cells resulting in the regulation of specific signalling pathways in recipient cells [3–6]. For instance, SkM-released exosome-like vesicles (ELVs) carry cell-type specific cargoes targeted for extracellular destinations (proteins [3–5] and micro [mi]RNAs [6]). It is now well-established that EVs modulate immune-regulatory processes, set up tumour escape mechanisms and mediate regenerative as well as degenerative processes [7–9]. In that context, we have shown that ELVs from differentiated muscle cells regulate myoblast proliferation and differentiation [5, 6]. Recently, we found that insulin resistance induced by lipid-enriched diet, was associated with an increase of SkM-released ELVs that were able to transfer the deleterious effects of palmitate between muscle cells [10]. These data supported the notion that SkM releases specific ELVs that act as ‘paracrine-like’ signals. Horizontal transfer of ELV cargoes has also been described in other insulin-sensitive tissues; for example, adipose tissue (crosstalk between small and large adipocytes [11]) and pancreas (transduction of apoptotic signals between beta cells [12]). Interestingly, we previously observed that C2C12-derived ELVs, i.v. injected into mice, were transferred to other tissues, strongly suggesting that SkM-derived ELVs could represent a mode of systemic communication [10]. As SkM is responsible for whole-body metabolism and energy homeostasis, determining whether muscle-released ELVs are involved in the development of insulin resistance is now emerging as a major research topic. In this study, we investigated whether SkM-released ELVs can be taken up by pancreas in vivo and in vitro and can deliver a functional cargo to beta cells. Furthermore, we determined whether this transfer was associated with the regulation of specific genes and affects the phenotype of the recipient beta cells. We found that ELVs isolated from diet-induced insulin-resistant muscle and normal muscle differed in terms of miRNA content and biological effects suggesting that the metabolic state of SkM affects ELV composition and action.
Methods
Mouse model
Male C57BL/6 mice (Harlan, Gannat, France) were housed in a common animal centre. Experiments were approved by the Local Board for Laboratory Animals (2010-008). Four-week-old mice were divided into two groups and fed either a standard chow diet (SCD: 57% carbohydrate, 5% fat and 18% protein) or an SCD enriched with 20% palm oil (HPD). After animals were killed, quadriceps were excised and used for ELV purification, and gene and protein expression analyses. The pancreas was enclosed in TissuTeck and frozen. Ten-micrometre sections were cut and stained with haematoxylin and eosin. Sections were observed with a Zeiss Axioskop2 microscope attached to a camera (Zeiss AxioCam ERc5s, Zeiss, Marly le Roi, France). No randomisation or blinding were performed. No data samples or animals were excluded from the study.
Mouse islet isolation
Proximal bile duct was cannulated and distended with collagenase type V (1 mg/ml, C9263, Sigma-Aldrich, Lyon, France) in Hanks’ balanced salt solution (HBSS) [13]. Pancreas was digested at 37°C for 10 min. The digested tissues were washed and filtered through a 500 μm mesh. Islets were purified by Ficoll gradient, and washed twice with HBSS solution.
Isolation and characterisation of muscle ELVs
Quadriceps were cut and incubated for 24 h in serum-free DMEM. Released particles were purified from these conditioned media. Cell debris and organelles were eliminated at 2,000g for 20 min and at 10,000g for 30 min. The supernatant fraction was filtered (0.22 μm filter). ELVs were pelleted by ultracentrifugation at 100,000g for 70 min at 4°C (Optima L-80-XP ultracentrifuge type 50-2Ti rotor, Beckman-Coulter, Villepinte, France). Pellets from a single sample were pooled, re-suspended in 25 ml PBS and again centrifuged at 100,000g for 70 min. Each pellet was finally re-suspended in 100 μl PBS. ELV proteins were quantified using the Bradford protein assay. Particle sizes were determined with NanoSight LM10 (Malvern, Orsay, France)
Cell lines
The murine insulin-secreting cell line MIN6B1 was provided by P. A. Halban (University of Geneva, Switzerland) with permission from J.-i. Miyazaki, University of Osaka, Japan [12, 14, 15]. They were cultured in DMEM–GlutaMAX medium (Invitrogen, Courtaboeuf, France) containing 25 mmol/l glucose and 4 mmol/l l-glutamine and supplemented with 15% FBS and 70 μmol/l β-mercaptoethanol 12,14-15. C2C12 mouse myoblasts (ATCC, Molsheim, France; CRL-1772) and 3T3-L1 pre-adipocytes (ATCC, CL-173) were grown in DMEM (25 mmol/l glucose) (Invitrogen) supplemented with 10% FBS, and 2 mmol/l l-glutamine. It should be noted that mycoplasma bacteria are around 100 nm in size and can present a potential problem for accurate exosome measurements. Thus, all the cells used in this study were tested and found to be negative for mycoplasma (MycoAlert™ Mycoplasma Detection Kit, Lonza, Amboise, France).
Production of C2C12-ELVs containing green fluorescent protein
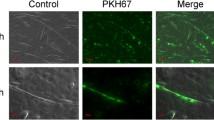
Unreplicative adenoviruses expressing green fluorescent protein (GFP) were generated as described previously [16]. C2C12 cells were infected with GFP-expressing adenovirus for 24 h in differentiation medium. Twenty-four hours later, all myotubes had green fluorescence in the cytoplasm indicating that all cells were infected. Myotubes were washed with PBS to remove non-integrated adenovirus and were incubated for another 48 h in ELV-depleted DMEM. GFP-containing ELVs, which had accumulated in conditioned medium for 48 h, were extracted. MIN6B1 or 3T3 cells (DMEM 10% FBS) were incubated with 2 μg GFP-ELVs per millilitre of medium [10]. Cells were visualised with a Zeiss Axiovert 200M Fluorescence/Live cell Imaging microscope equipped with the Axiovision software (Zeiss).
Total RNA extraction from ELVs
Total RNA was extracted from ELVs by using TriPure Isolation Reagent (Roche Applied Science, Meylan, France). RNA concentration was measured with a Nanodrop spectrophotometer, ND-1000 (Thermo-Fisher, Waltham, MA, USA).
miRNA profiling by qRT-PCR quantification
Quantification of ELV miRNAs was determined using quantitative real-time RT-PCR (qRT-PCR)-based 384-well TaqMan low-density array cards (Applied Biosystems, Saint Aubin, France). Total RNA was reverse-transcribed using TaqMan miRNA Reverse Transcription Kit in combination with the stem–loop Megaplex primers in 7.5 μl. Next, 2.5 μl of the Megaplex reverse transcription product was mixed with 2.5 μl of Megaplex PreAmp Primers and 12.5 μl TaqMan PreAmp Master Mix in a 25 μl PCR reaction and pre-amplified. The pre-amplified cDNA was then diluted with 0.1× TE buffer (pH 8.0) to 100 μl. The diluted cDNA product (9 μl) was used for qRT-PCR runs. qRT-PCR was performed on a 7900HT thermocycler (Applied Biosystems) using the manufacturer’s recommended cycling conditions. Ct values were calculated with the SDS software using automatic baseline settings with assigned minimum Ct threshold of 0.2. Ct values >40 were excluded from data analysis. Each card included three miRNA endogenous controls and one miRNA assay not related to mouse. For each card, quality controls were performed on the raw data by checking internal controls and using box plot diagrams. We used the mean expression level of all fully observed miRNAs for normalisation [17]. Group comparisons were made by using the Student’s t test (p < 0.05) on normalised data, to select the differentially expressed miRNAs.
Microarray data analysis
RNA profiling in pancreatic islets or MIN6B1 cells was performed using a Mouse GE 4×44K v2 Microarray kit (Agilent Technologies, Les Ulis, France). Total RNA (100 ng) was labelled with Cy3 using the Low Input Quick Amp Labelling kit from Life Technologies (St-Aubin, France) and microarrays were hybridised and scanned following the manufacturer’s instructions. Data quality analysis and normalisation were performed with R environment using Agi4×44PreProcess and Limma packages [18]. Datasets are available from GEO database (GSE59429 and GSE67199).
Effect of muscle-release ELVs on MIN6B1 cells and mouse islet proliferation
MIN6B1 cells, cultured on poly-l-lysine-coated glass cover slips and treated for 24 h with standard diet-ELVs (SD-ELVs) or HPD-ELVs, were fixed with methanol and permeabilised with 0.5% (wt/vol.) saponin (Sigma-Aldrich). Isolated islets, cultured on collagen-coated glass cover slips and treated for 48 h with ELV-SD or ELV-HP, were fixed with paraformaldehyde and permeabilised with TritonX 100 0.1%. Cover slips were incubated with antibodies against Ki67 (15580, 1/500, Abcam, Paris, France) and insulin (A0564, 1/100, Dako Agilent Technologies) and then with anti-rabbit Alexa-Fluor-488 (A11034, 1/1000, Invitrogen) and anti-guinea pig Alexa-Fluor-546 antibodies (A11074, 1/1000, Invitrogen). At the end of incubations, cover slips were washed with PBS and mounted with DAPI containing VECTASHIELD Antifade Mounting Medium (Vector Laboratories, Peterborough, UK) and three images of at least 100–300 cells per condition (islets) or 1 × 103 cells (MIN6B1) were collected using a fluorescence microscope.
Transfection assays
Transient transfections were performed with 5 μl of ExGen500 for 1 μg of DNA (EUROMEDEX, France). MIN6B1 cells were plated on 6-well plates. At 60–70% confluence, cells were transfected with 1 μg of pEZX-MT01 vector containing the 3′-UTR segment of Ptch1 (GeneCopoeia, LabOmics, Nivelles, Belgium). After 18 h, MIN6B1 cells were incubated with serum-free DMEM containing either SD-ELVs or HPD-ELVs for 24 h. Firefly luciferase activities were normalised with Renilla luciferase activities to minimise experimental variabilities caused by differences in cell viability or transfection efficiency (Dual-Luciferase Reporter Assay System; Promega, Charbonnières-les-Bains, France).
To validate the binding of miR-16 to Ptch1 3′-UTR region, HEK293T cells (ATCC, CRL-11268) with low endogenous expression of miR-16, were co-transfected with 200 ng pEZX-MT01 vector containing the 3′-UTR segment of Ptch1 and either 800 ng of miR-16 precursors (pEZX-MR04 vectors) (GeneCopoeia) or 800 ng scramble non-targeting control (miR-CTRL), or 30 pmol of anti-miR-16 (Ambion Anti-miR miRNA inhibitors, LifeTechnologies). After 48 h, luciferase activity was quantified. Modulation of Ptch1 expression by miR-16 was validated by transfecting MIN6B1 with miR-16 precursors or anti-miR-16, or respective controls. Involvement of Ptch1 in beta cell proliferation was confirmed by transfecting MIN6B1 cells with 25 nmol/l siRNA against Ptch1 for 48 h (Qiagen, Les Ulis, France).
qRT-PCR
qRT-PCR was performed using ABsolute QPCR SYBR Green ROX Mix (Abgene, Courtaboeuf, France) with a Rotor-Gene 6000 system (Corbett Life Science, Qiagen).
Biodistribution of ELVs
ELVs were pelleted in the presence of 1 μmol/l fluorescent lipophilic tracer (1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide [DiR]) (D12731, Thermo Fisher/Life Technologies, Paisley, UK) by ultracentrifugation (110,000 g, 90 min). The near infrared fluorescent lipophilic DiR is weakly fluorescent in water, but highly fluorescent and quite photostable when incorporated into membranes. It has excitation and emission maxima in the near infrared region where many tissues are optically transparent. DiR-labelled ELVs were injected either through the tail vein or intramuscularly (i.m.) into the right tibialis anterior (r.TA) of female NMRI mice (Scanbur, Sollentuna, Sweden). As a control we injected with mock-PBS-DiR, performed the same as the labelling of ELVs but without ELVs. After 24 h animals were killed and the organs were carefully harvested and imaged for 2 seconds (excitation 710, emission 760) using a high sensitive CCD camera IVIS Spectrum (PerkinElmer, Upplands Väsby, Sweden). Animal experiments were approved by the Swedish Local Board for Laboratory Animals and designed to minimise animal suffering and pain.
Statistical analysis
Results are expressed as mean ± SEM. Student’s t test was used to analyse the difference between SCD and HPD groups. A p value <0.05 was considered as statistically significant.
Results
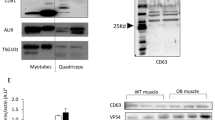
As previously shown [10], mice fed with HPD for 16 weeks have a higher body weight, are glucose-intolerant and insulin-resistant compared with mice fed with SCD (Electronic Supplementary Material [ESM] Fig. 1). EVs released from HPD or SCD quadriceps were prepared ex vivo from conditioned media by ultracentrifugation [10, 19]. We analysed the size distribution of the whole population of secreted vesicles with nanoparticle tracking analysis technique [20] and transmission electron microscopy (TEM). Vesicle size distributions displayed bell-shaped curves with a mean size of 93 nm (SD-ELVs) and 110 nm (HPD-ELVs) indicating that HPD does not affect the size of muscle-released vesicles (Fig. 1a). Size of the vesicles and the fact that many of them expressed CD81 and CD63 (Fig. 1b, c) indicated that the vesicle pellet was composed mainly of exosomes [21]. Because there is still a lack of consensus about the isolation techniques to accurately separate subpopulations of EVs of the same size [22], hereafter we use the term of ELVs

Characterisation of quadriceps-released ELVs. (a) Vesicle sizes from quadriceps of SCD or HPD mice. (b) TEM of CD81 immunolabelled quadriceps-released ELVs. Scale bar, 100 nm. (c) Detection of CD63 in muscle ELVs by western blot. Antibodies have been described and thoroughly validated previously [5, 6, 10]
Muscle-released ELVs are incorporated into pancreas in vivo
Two preparations of DiR-stained SD-ELVs or HPD-ELVs were injected into mice tail veins [23]. Twenty-four hours post-injection, fluorescence was predominant in the spleen followed by the liver and the lungs (ESM Fig. 2). Both types of ELVs (SCD vs HPD) accumulated in the same tissues. Subsequently we speculated that in vivo, SkM-ELVs could be released into blood and target other organs. However, because specific muscle-markers at the surface of mouse SkM-ELVs have not yet been identified, it is presently impossible to follow their release from SkM to the blood. Therefore, labelled ELVs were injected i.m into the r.TA and their biodistribution was analysed 24 h post-injection. In parallel, to exclude contamination of blood with labelled ELVs during i.m. injection, the same quantity of labelled ELVs was i.v.-injected in another group of animals. As shown in Fig. 2a, the global distribution of the i.m.-injected ELVs was different from that of the i.v.-injected ELVs, with less accumulation in liver and spleen and more in pancreas, muscle, kidneys and gastrointestinal (GI)-tract. These data confirmed that ELVs do not escape into blood during i.m. injection and suggested that muscle-released ELVs can target other insulin-sensitive tissues as well as the pancreas. In addition, these data also confirmed the paracrine-like action of ELVs [10] as fluorescence was also detected in the right quadriceps and in the untreated left tibialis anterior (Fig. 2b).

Muscle-released ELVs can accumulate in pancreas. To obtain enough ELVs to repeat injections, we have chosen to use HEK293-released ELVs. These cells produce more ELVs than muscle cells and their ELVs have the same biodistribution as muscle ELVs [33]. Labelled ELVs were injected i.m. (n = 3) into r.TA and compared with i.v. (n = 4) injections. Each animal was injected with 2 × 1011 particles, dissolved in PBS (200 μl for i.v., 50 μl for i.m.). Measurements were normalised to background by subtracting the fluorescence values of the organs with that of the PBS-treated organs. (a) Global distribution comparing i.v. (black bars) vs i.m. (grey bars); *p < 0.05, **p < 0.01 for i.m. vs i.v. (b) Graph showing values of fluorescence of the muscles (r.TA, left tibialis anterior [l.TA] and right [r.] quadriceps) for i.m. (c) MIN6B1 or 3T3-L1 cells incubated with GFP-containing C2C12-released ELVs [5]. (d) Quantification of miR-206 in MIN6B1 cells treated with 2 μg of ELVs from C2C12 myotubes (n = 3); AU, arbitrary units; ***p < 0.001 for ELV-treated cells vs untreated cells
Myotube-released ELVs transfer their cargo into beta cells
To further demonstrate that muscle-released ELVs can transfer their content into other cell types, GFP-containing ELVs accumulated in conditioned medium from C2C12 were used to treat either MIN6B1 or 3T3 cells. After 24 h, fluorescent cells were detected indicating that muscle-released ELVs had transferred their GFP content into beta cells or 3T3 adipocytes (Fig. 2c). In addition, increased levels of the muscle-specific miR-206 was detected in MIN6B1 cells (Fig. 2d), demonstrating that myotube-ELVs also horizontally transferred this miRNA into beta cells. Fluorescence was also detected in exocrine pancreas and endothelium, post i.v injection of myotube-released ELVs, suggesting that muscle ELVs can also transfer their content to other cell types of the pancreas (ESM Fig. 3).
SD-ELVs and HPD-ELVs differentially regulate gene expression in ΜΙΝ6Β1 cells
Then we wondered whether HPD-ELVs induced specific transcriptional signatures in MIN6B1 cells as previously observed in muscle cells [10]. Microarray analyses showed that the level of 460 mRNAs was significantly changed in response to HPD-ELVs compared with SD-ELVs (ESM Table 1). Modulated transcripts mainly encoded proteins for membrane receptors, transcription factors, enzymes, signalling pathways and proteasome/proteases. Interestingly, HPD-ELVs modulated a significant number of genes involved in immune response, a function affected in pancreatic islets of patients with type 2 diabetes [24] (ESM Table 1).
To determine whether miRNAs from muscle ELVs might be involved in this transcriptional signature change in MIN6B1 cells, the 3′-UTR regions of the regulated genes were analysed with DIANA-mirExTra, an algorithm that identifies miRNA effects on the expression levels of protein-coding transcripts based on the frequency of six-nucleotide-long motifs (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=site/index), accessed October 2015. Sixty-four and 93 motifs corresponding to binding sites for 51 and 70 miRNAs were enriched in the 3′-UTR regions of the up- and downregulated genes, respectively. Among the 70 predicted miRNAs, eight were included in muscle-released ELVs, including two myomiRs (miR-1 and miR-206) and one significantly more abundant in HPD-ELVs compared with SD-ELVs (miR-16) estimated by the mean of Ct values after normalisation (Table 1). Among the 51 predicted miRNAs, nine were included in muscle ELVs but none was differentially expressed in SD-ELVs vs HPD-ELVs. Taken altogether, these data suggested that miRNAs from HPD-ELVs contributed to the downregulation of the expressions of 160 genes in MIN6B1 recipient cells.
HPD-ELVs miRNAs modulate the level of Ptch1 in MIN6B1 cells
As combinations of different miRNA binding sites have synergistic effects on gene expression [25], we further focused on the downregulated genes targeted by multiple HPD-ELV miRNAs. As shown in Table 2, the top ten targeted genes were mainly involved in cell cycle and proliferation. In agreement with this result, MIN6B1 cells treated for 24 h with HPD-ELVs had increased proliferation rate compared with cells exposed to SD-ELVs (Fig. 3a), without modifications of insulin secretion (data not shown). This result was reproduced with isolated islets (Fig. 3b, c). In vivo, HPD mice were hyperinsulinaemic after 6 h fasting (ESM Fig. 1) and their pancreatic islets showed increased size (Fig. 3d) and altered gene expressions of a significant number of genes involved in proliferation and development (ESM Table 2). Taken together, these data showed that HPD-induced insulin resistance had a deep impact on islet size in vivo and suggested that this result could be partially reproduced in vitro when beta cells were treated with HPD-ELVs but not with SD-ELVs.

Muscle-released ELVs modulate beta cell proliferation and regulate Ptch1. MIN6B1 cells (a) or isolated islets (b) were treated with SD-ELVs or HPD-ELVs for 48 h. Results are expressed as percentage of Ki67-positive nuclei vs total number of nuclei. (c) Immunolabelling of isolated islets treated with SD- or HPD-ELVs; Blue, nuclei; Green, Kif67-positive nuclei; Red, insulin. (d) Histology of islets of Langerhans (n = 3 mice); scale bar, 50 μm. (e) Ptch1 expression in islets of SCD vs HPD mice. (f) Luciferase activity of the construct containing the Ptch1 3′-UTR region, in transfected MIN6B1 cells treated with SD- or HPD-ELVs. (g) MiR-16 expression in MIN6B1 cells treated with SD- or HPD-ELVs. (h) Luciferase activity of the Ptch1 3′-UTR region construct, co-transfected with: (1) pre-miR control; or (2) pre-miR-16. Only the pre-miR-16 condition showed a significant reduction of luciferase activity. Co-transfection with an anti-miR-16 (3) reduced the binding of miR-16 on Ptch1. (i) Ptch1 expression in MIN6B1 cells treated with miR-16 or pre-miR-16. (j) Number of MIN6B1 nuclei pre-treated with siRNA against Ptch1. (k) MIN6B1 cells treated with siRNA against Ptch1 (magnification ×10). Ctl, control. *p < 0.05, **p < 0.01 for all figure parts as shown
Among the ten genes highly targeted by miRNAs from HPD-ELVs in MIN6B1, we focused on Ptch1, a receptor involved in the sonic hedgehog pathway, suggested to play an important role during pancreas development and disease [26]. This gene was also downregulated at the mRNA level in islets from HPD-fed animals vs SCD (Fig. 3e). In addition, the 3′-UTR region of Ptch1 had a predicted binding site for miR-16, which was more abundant in HPD-ELVs than in SD-ELVs (Table 1). To validate that HPD-ELV miRNAs could collectively regulate gene expression in MIN6B1 cells, we performed luciferase assays with the full-length 3′-UTR construct of Ptch1. As shown in Fig. 3f, Ptch1 3′-UTR luciferase activity was significantly reduced when MIN6B1 cells were treated with HPD-ELVs, while addition of SD-ELVs at the same concentration had no effect. This result was corroborated by an increase of miR-16 levels (Fig. 3g), further confirming the transfer of miRNAs from muscle ELVs into beta cells. The binding of miR-16 to the Ptch1 3′-UTR region was validated in a co-transfection assay (Fig. 3h), and modulation of miR-16 concentration resulted in regulation of Ptch1 mRNA level, suggesting that this miRNA likely participated in the effect of HPD-ELVs on the downregulation of Ptch1 in beta cells. Finally, reducing Ptch1 level led to an increase of MIN6B1 proliferation (Fig. 3j, k) confirming the link between Ptch1 and cell proliferation.
Discussion
Palmitic acid, the most abundant saturated NEFA of palm oil, has received considerable attention as it is suspected to participate in the onset of metabolic diseases [27]. Indeed, mice fed with HPD for 16 weeks developed hyperglycaemia, hyperinsulinaemia, glucose-intolerance and insulin resistance. They displayed larger islet size compared with SCD animals and altered expressions of genes involved in immune response. We previously showed that HPD also had a deep impact on muscle homeostasis and resulted in an increased release of SkM-ELVs [10]. Moreover, we showed that SkM-released ELVs horizontally transfer between muscle cells signals mediating the deleterious effects of palmitate [10]. Until now, the relationship between insulin resistance in SkM and decrease of insulin secretion by the pancreas remains an enigma [28]. For example, it was found that muscle-specific peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) knockout mice, with abnormal glucose homeostasis in SkM but normal peripheral insulin-sensitivity, had expanded beta cell mass and increased expression of several pro-inflammatory genes suggesting a SkM-beta cell crosstalk [29]. In this study, we suggest that muscle-released ELVs might participate in these molecular dialogues. Some data have shown that ELVs have a distinct protein and lipid composition resembling that of the cell of origin [30] suggesting that ELVs may preferentially interact with their parent cells. However, because there is not enough proteomic and lipidomic data from EVs released from all mouse tissues, it is impossible to design antibodies to isolate specifically SkM-released ELVs from blood and validate this hypothesis. To try to bypass this technical issue, we have loaded SkM-ELVs with GFP and demonstrated that they can be incorporated into beta cells, in vitro. More importantly, we have demonstrated that either injected i.v. or i.m., SkM-derived ELVs are incorporated into pancreas in vivo. However, it has to be mentioned that this in vivo incorporation might reflect uptake and re-release of ELVs from various cell types before reaching pancreas. Indeed, it was previously found that ELVs declined quickly in serum after injection and that macrophages are involved in the clearance [31–33]. Moreover injected ELVs generally distribute to organs of the mononuclear phagocyte system with highest accumulation in the liver, followed by spleen, GI-tract and lungs [34]. Interestingly, labelled ELVs injected either i.m. or i.v. rendered different distributions. It is likely that ELVs injected intramuscularly are incorporated directly into myotubes and then are released as muscle-released ELVs. Therefore, these data suggest that in addition to a route involving patrolling macrophages, there might be another possible direct route for ELVs incorporation into organs.
We further demonstrated that, at least in vitro, ELVs delivered functional cargoes as the levels of myomiRs were increased in MIN6B1 cells treated with muscle ELVs. MiRNA profiling of HPD- vs SD-ELVs showed that insulin resistance in SkM was associated with the release of ELVs containing specific miRNAs (increased miR-16 level in HPD-ELVs). Presently, it is not clear how miRNA export is regulated. Among possible mechanisms [35–38] it was suggested that the ceramide pathway may be involved [39]. This result suggests that de novo synthesis of ceramides from palmitate in SkM during HPD [40] could contribute to modify the population of exported miRNAs. Accordingly, we found that HPD muscle-released ELVs affected proliferation and gene expression in beta cells and bioinformatic analyses strongly suggested that transferred miRNAs from HPD-ELVs might be involved in these regulations. We confirmed this hypothesis by focusing on Ptch1 as a reporter gene to validate that muscle-released ELVs were indeed able to communicate with MIN6B1 cells through the in vitro transfer of miR-16 enriched in HPD-ELVs. Ptch1 of the hedgehog pathway was downregulated both in HPD-SkM-ELVs-treated MIN6B1 cells and in hypertrophic pancreatic islets of HPD-fed mice and was predicted to be a miR-16 target gene. The glucose-intolerance phenotype observed in Ptch1 mutant mice [41] and the requirement for hedgehog signals for insulin transcription and secretion in insulinoma cells [42] indicate that this pathway might be considered as a potential factor in the pathogenesis of type 2 diabetes. In addition, it was demonstrated that PTCH1 is involved in the inhibition of cell proliferation [43–45] and we confirmed this data in beta cells. Of note, although Ptch1 was also downregulated in islets from HPD vs SCD-fed animals and in MIN6B1 treated with HPD-ELVs, its regulation by miR-16 could not be firmly confirmed in vivo as miR-16 was not concomitantly upregulated in these islets. This result suggests that miR-16 upregulation would occur only in beta cells, in vivo. In addition to miRNAs, ELVs carry also numerous proteins, and nucleic acids that can collectively affect multiple signalling pathways inside targeted cells and the specific contribution of each component is still unknown. ELVs can also transfer lipids, such as eicosanoids, fatty acids and cholesterol [46], and we demonstrated that muscle-released ELVs are also able to transfer palmitate and oleate between muscle cells [10].
These data suggest that SkM-released ELVs might transmit signals to the pancreas during the development of insulin resistance. The fact that HPD-ELVs had no effect on beta cell insulin secretion indicates that beta cells retained a fully differentiated phenotype. Thus, SkM-derived ELVs, together with nutrients and hormones, might contribute to adaptations in beta cell mass occurring during insulin resistance. However, further studies are needed to validate this concept and caution must be exercised not to over-interpret these data until a more physiologically appropriate model system is tested. Indeed, the generation of transgenic mice expressing GFP in SkM-released EVs would help to quantify in vivo the impact of SkM-released EVs in the crosstalk between SkM and key metabolic organs during the development of metabolic diseases.
Abbreviations
- DiR:
-
1-Dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide
- ELVs:
-
Exosome-like vesicles
- EVs:
-
Extracellular vesicles
- GFP:
-
Green fluorescent protein
- HBSS:
-
Hanks’ balanced salt solution
- HPD:
-
High palmitate diet
- HPD-ELVs:
-
High palmitate diet-ELVs
- GI:
-
Gastrointestinal
- i.m:
-
Intramuscular
- miRNA:
-
MicroRNA
- qRT-PCR:
-
Quantitative real-time RT-PCR
- r.TA:
-
Right tibialis anterior
- SCD:
-
Standard chow diet
- SD-ELVs:
-
Standard diet-ELVs
- SkM:
-
Skeletal muscle
- SkM-ELVs:
-
Skeletal muscle-ELVs
- TEM:
-
Transmission electron microscopy
References
Bouzakri K, Plomgaard P, Berney T et al (2011) Bimodal effect on pancreatic beta-cells of secretory products from normal or insulin-resistant human skeletal muscle. Diabetes 60:1111–1121
Pedersen BK, Febbraio MA (2012) Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev 8:457–465
Guescini M, Guidolin D, Vallorani L et al (2010) C2C12 myoblasts release micro-vesicles containing mtDNA and proteins involved in signal transduction. Exp Cell Res 316:1977–1984
Le Bihan MC, Bigot A, Jensen SS et al (2012) In-depth analysis of the secretome identifies three major independent secretory pathways in differentiating human myoblasts. J Proteome 21:344–356
Forterre A, Jalabert A, Berger E et al (2014) Proteomic analysis of C2C12 myoblast and myotube exosome-like vesicles: a new paradigm for myoblast-myotube cross talk? PLoS One 9, e84153
Forterre A, Jalabert A, Chikh K et al (2014) Myotube-derived exosomal miRNAs downregulate Sirtuin1 in myoblasts during muscle cell differentiation. Cell Cycle 13:78–89
Lopez-Verrilli MA, Court FA (2013) Exosomes: mediators of communication in eukaryotes. Biol Res 46:5–11
Thery C, Boussac M, Veron P et al (2001) Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol 166:7309–7318
Mathivanan S, Ji H, Simpson RJ (2010) Exosomes: extracellular organelles important in intercellular communication. J Proteome 73:1907–1920
Aswad H, Forterre A, Wiklander OP et al (2014) Exosomes participate in the alteration of muscle homeostasis during lipid-induced insulin resistance in mice. Diabetologia 57:2155–2164
Muller G, Schneider M, Biemer-Daub G et al (2011) Upregulation of lipid synthesis in small rat adipocytes by microvesicle-associated CD73 from large adipocytes. Obesity 19:1531–1544
Guay C, Menoud V, Rome S et al (2015) Horizontal transfer of exosomal microRNAs transduce apoptotic signals between pancreatic beta-cells. Cell Commun Signal 13:17
Mai G, Bucher P, Morel P et al (2005) Anti-CD154 mAb treatment but not recipient CD154 deficiency leads to long-term survival of xenogeneic islet grafts. Am J Transplant 5:1021–1031
Lilla V, Webb G, Rickenbach K et al (2003) Differential gene expression in well-regulated and dysregulated pancreatic beta-cell (MIN6) sublines. Endocrinology 144:1368–1379
Miyazaki J, Araki K, Yamato E et al (1990) Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127:126–132
Dif N, Euthine V, Gonnet E et al (2006) Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J 400:179–188
Deo A, Carlsson J, Lindlof A (2011) How to choose a normalization strategy for miRNA quantitative real-time (qPCR) arrays. J Bioinforma Comput Biol 9:795–812
Smyth GK (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology 3: Article3
Thery C, Amigorena S, Raposo G (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology / editorial board, Juan S Bonifacino Chapter 3: Unit 3 22
Dragovic RA, Gardiner C, Brooks AS et al (2011) Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine 7:780–788
Lotvall J, Hill AF, Hochberg F et al (2014) Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 3:26913
Bobrie A, Colombo M, Krumeich S et al (2012) Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation. J Extracell Vesicles 16:1
Ohno S, Takanashi M, Sudo K et al (2012) Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther 21:185–191
Esser N, Legrand-Poels S, Piette J et al (2014) Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract 105:141–150
Granjon A, Gustin MP, Rieusset J et al (2009) The microRNA signature in response to insulin reveals its implication in the transcriptional action of insulin in human skeletal muscle and the role of a sterol regulatory element-binding protein-1c/myocyte enhancer factor 2C pathway. Diabetes 58:2555–2564
Lau J, Kawahira H, Hebrok M (2006) Hedgehog signaling in pancreas development and disease. Cell Mol Life Sci 63:642–652
Vega-Lopez S, Ausman LM, Jalbert SM, Erkkila AT, Lichtenstein AH (2006) Palm and partially hydrogenated soybean oils adversely alter lipoprotein profiles compared with soybean and canola oils in moderately hyperlipidemic subjects. Am J Clin Nutr 84:54–62
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl 2):S157–S163
Handschin C, Choi CS, Chin S et al (2007) Abnormal glucose homeostasis in skeletal muscle-specific PGC-1α knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. JCI 117:3463–3474
Kalra H, Simpson RJ, Ji H, Aikawa E et al (2012) Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol 10, e1001450
Deng ZB, Poliakov A, Hardy RW et al (2009) Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58:2498–24505
Takahashi Y, Nishikawa M, Shinotsuka H et al (2013) Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol 165:77–84
Imai T, Takahashi Y, Nishikawa M et al (2015) Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J Extracell Vesicles 4:26238
Wiklander OP, Nordin JZ, O'Loughlin A et al (2015) Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles 4:26316
Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F et al (2013) Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4:2980
Gibbings DJ, Ciaudo C, Erhardt M et al (2009) Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol 11:1143–1149
Squadrito ML, Baer C, Burdet F et al (2014) Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep 8:1432–1446
Koppers-Lalic D, Hackenberg M, Bijnsdorp IV et al (2014) Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep 8:1649–1658
Kubota S, Chiba M, Watanabe M et al (2015) Secretion of small/microRNAs including miR-638 into extracellular spaces by sphingomyelin phosphodiesterase 3. Oncol Rep 33:67–73
Hage Hassan R, Bourron O, Hajduch E (2014) Defect of insulin signal in peripheral tissues: important role of ceramide. World J Diabetes 5:244–257
Hebrok M, Kim SK, St Jacques B et al (2000) Regulation of pancreas development by hedgehog signaling. Development 127:4905–4913
Thomas MK, Rastalsky N, Lee JH, Habener JF (2000) Hedgehog signaling regulation of insulin production by pancreatic beta-cells. Diabetes 49:2039–2047
Trazzi S, Mitrugno VM, Valli E et al (2011) APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum Mol Genet 20:1560–1573
Rudin CM (2012) Vismodegib. Clin Cancer Res 18:3218–3222
Jenkins D (2009) Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal 21:1023–1034
Record M, Carayon K, Poirot M, Silvente-Poirot S (2014) Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim Biophys Acta 1841:108–120
Acknowledgements
We thank E. Errazuriz (Centre Commun d’Imagerie de Laënnec, SFR Santé Lyon-Est, University of Lyon, France) for TEM images, and A-M Madec from CarMeN laboratory, for her help in growing MIN6B1 cells. We thank V. Euthine, C. Durand and S. Chanon, from CarMeN laboratory, for their help in cell culture and immunostaining, and E. Lefai for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Fondation pour la Recherche Médicale (DRM20101220456), and by a grant from the biopharmaceutical company AstraZeneca (SR) and by the Swiss National Science Research Foundation (No. 310030-146138) (RR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All the authors provided substantial contributions to the conception and design, acquisition and analysis of the data. All the authors have participated in drafting the article and approved the version to be published. SR is responsible for the integrity of the work as a whole.
Additional information
Audrey Jalabert and Guillaume Vial contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 91 kb)
ESM Fig. 2
(PDF 13 kb)
ESM Fig. 3
(PDF 307 kb)
ESM Table 1
(XLS 162 kb)
ESM Table 2
(PDF 26 kb)
Rights and permissions
About this article

Cite this article
Jalabert, A., Vial, G., Guay, C. et al. Exosome-like vesicles released from lipid-induced insulin-resistant muscles modulate gene expression and proliferation of beta recipient cells in mice. Diabetologia 59, 1049–1058 (2016). https://doi.org/10.1007/s00125-016-3882-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3882-y