
Abstract
Aims/hypothesis
T cells play a major role in the pathogenesis of type 1 diabetes, and there is great interest in developing curative immunotherapies targeting these cells. In this study, a monoclonal antibody (mAb) targeting the T cell receptor β-chain (TCRβ) was investigated for its ability to prevent and reverse disease in mouse models of diabetes.
Methods
RIP-OVAhi (C57BL/6-Tg(Ins2-OVA)59Wehi/WehiJ) mice adoptively transferred with ovalbumin-specific T cells (an induced model of diabetes) and NOD mice (a spontaneous model of diabetes) were used to test anti-TCRβ mAb therapy as a means of preventing and reversing type 1 diabetes.
Results
A single dose of anti-TCRβ completely prevented disease in RIP-OVAhi mice without inducing the release of inflammatory cytokines. Transient anti-TCRβ therapy prevented diabetes in 90% of NOD mice and reversed the disease after its onset in 73% of NOD mice. Long after the remission of type 1 diabetes, the anti-TCRβ treated mice were able to reject BALB/c skin allografts with normal kinetics while maintaining normoglycaemia. Treatment did not cause significant reductions in lymphocyte numbers in the spleen or pancreatic lymph nodes, but did result in a decreased percentage of chemokine receptor 9 (CCR9) positive, CD8+ T cells. Notably, anti-TCRβ therapy increased the expression of programmed death 1 (PD-1) on the surface of the T cells; PD-1 expression is important for maintaining anti-TCRβ-induced self-tolerance, as type 1 diabetes recurs in mice following a blockade of PD-1 signalling.
Conclusions/interpretation
Anti-TCRβ mAb is a safe and effective immunotherapy that results in reduced numbers of CCR9+ T cells, an increased expression of PD-1 on T cells and the restoration of self-tolerance in NOD mice.
Similar content being viewed by others

Introduction
Globally, the incidence of type 1 diabetes is increasing, with 30,000 new diagnoses each year in the USA and a trend towards a 70% increase in prevalence among European children by 2020 [1]. A combination of genetic and environmental factors is thought to contribute to the autoimmune response against the insulin-producing beta cells of the pancreas, mediated in large part by T cells [2]. Many reports in humans and rodent models demonstrate the critical role of CD4+ and CD8+ T cells in the pathogenesis of type 1 diabetes [3, 4] and have influenced the design of curative immunotherapies. Recent advances have allowed the generation of functional pancreatic beta cells from human pluripotent stem cells [5]; however, an effective immunotherapeutic approach is needed to protect these newly generated insulin-producing cells from autoimmune attack.
Although many immune interventions prevent disease in the NOD mouse model of diabetes [6], no treatments are currently approved for clinical use in preventing type 1 diabetes [7]. Fewer therapies have been successful in reversing type 1 diabetes in NOD mice, notably, recombinant GAD65 autoantigen [8], low dose IL-2 [9] and anti-CD3 monoclonal antibody (mAb) [10]. These therapies have been difficult to translate into clinical benefits for patients with type 1 diabetes [11]. Recent reports from clinical trials of new-onset type 1 diabetes have shown limited efficacy compared with placebo with GAD-alum [12–15], low-dose IL-2 plus rapamycin [16] or anti-CD3 mAb therapies [17–19]. Post hoc analyses in some of these trials have demonstrated clinical efficacy in select patient populations. Other trials, including different formulations of anti-CD3 and alternative T cell-targeting agents, have shown promise and continue to enrol new patients [15, 20, 21]. Some clinical trials, such as those testing abatacept (a CTLA4–Ig fusion protein) have demonstrated success in humans despite failing in NOD mice [14]. Regardless of the difficulty of translating these approaches from mouse to human, continued research into the identification of novel immune therapeutics to reverse type 1 diabetes in NOD mice is warranted as this remains the most useful preclinical model. Furthermore, a deeper understanding of cellular and molecular mechanisms in NOD mice may help to design more successful clinical trials.
A number of anti-TCR mAbs have also been tested for prolonging allograft survival, including clones T10B9 [22] and BMA031 [23] in humans and H57-597 in mice [24]. Recently, our group demonstrated the efficacy of an anti-T cell receptor β-chain (TCRβ) mAb (H57-597) in inducing long-term cardiac allograft survival in mice through a regulatory T cell (Treg)-dependent mechanism [25]. This same clone has been effective in preventing type 1 diabetes in NOD mice [26]; however, the mechanism of action and demonstration of long-term, durable self-tolerance have not been described. This work demonstrates the efficacy and safety of anti-TCRβ mAb treatment in restoring durable self-tolerance in diabetic NOD mice and provides unique insights into the mechanism underlying its ability to reverse type 1 diabetes in NOD mice.
Methods
Mice
Wild-type (WT) C57BL/6, WT BALB/c, C57BL/6-Tg(Ins2-OVA)59Wehi/WehiJ (RIP-OVAhi), NOD/ShiLtJ (NOD) and NOD.Cg-Prkdc scidIl2rgtm1Wjl/SzJ (NOD-scid) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). B6.129S7-Rag1 tm1MomTg(TcraTcrb)425Cbn (Rag1 −/−OT-II) and B6.129S7-Rag1 tm1MomTg(TcraTcrb)1100Mjb (Rag1 −/−OT-I) mice were obtained from Taconic Farms (Hudson, NY, USA). The animals were maintained at the University of Toledo specific pathogen-free or sterile animal facility according to institutional guidelines. Animal work was performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council.
Reagents
All fluorescence-conjugated mAbs, including anti-CD4 (GK1.5), anti-CD8a (Ly-2), anti-chemokine receptor 9 (CCR9; eBioCW-1.2) and anti-programmed death 1 (PD-1; J43), were purchased from eBioscience (San Diego, CA, USA). Purified anti-TCRβ (H57-597) mAb, anti-CD3 (145-2C11) mAb, anti-programmed 1 death ligand (PDL1; 10F.9G2) mAb and isotype (hamster IgG Ab) were obtained from Bio X-Cell (West Lebanon, NH, USA). Ovalbumin (OVA)323–339 peptide was obtained from Ohio Peptide (Powell, OH, USA).
Blood glucose measurements
All blood glucose measurements were performed using a ReliOn Ultima glucose meter (Abbott Diabetes Care, Alameda, CA, USA). The diagnosis of terminal diabetes in the prevention experiments was made after two consecutive days of blood glucose readings over 27.8 mmol/l, and new-onset diabetes was defined as two consecutive days of readings between 13.9 and 19.4 mmol/l for the remission experiments.
Acute type 1 diabetes in RIP-OVAhi mice
The RIP-OVAhi mice were adoptively transferred with 5 × 105 purified OT-I cells from Rag1 −/−OT-I mice, 1 × 106 purified OT-II cells from Rag1 −/−OT-II mice and 2 × 105 bone marrow-derived dendritic cells (DCs) pulsed with 10 μg/ml OVA323–339 peptide. Recipient mice (n = 4 per group) then received i.p. injections of either sterile PBS or a single dose of 1 mg/kg anti-TCRβ and were monitored for their blood glucose concentrations as indicated.
Serum cytokine measurements
WT C57BL/6 mice received i.p. injections of anti-CD3, anti-TCRβ or isotype at doses of 0.1, 1 or 5 mg/kg as indicated. Normoglycaemic and new-onset diabetic NOD mice received i.p. injections of 1 mg/kg anti-CD3, anti-TCRβ or isotype. At different times after injection, serum samples were isolated and analysed for IL-2, IL-6, IFN-γ and TNF-α levels using quantitative mouse Duoset ELISA kits (R&D Systems, Minneapolis, MN, USA).
Monitoring and treatment of type 1 diabetes in NOD mice
To prevent type 1 diabetes, 8-week-old female NOD mice received once-weekly i.p. injections for 4 weeks of 1 mg/kg anti-CD3, anti-TCRβ or isotype, and their blood glucose levels were monitored until they were 40 weeks of age. To investigate the remission of type 1 diabetes, NOD mice with new-onset diabetes received i.p. injections daily for 10 days with 1 mg/kg isotype, anti-TCRβ or anti-CD3. The blood glucose level was monitored for 80 days after the onset of type 1 diabetes to detect any recurrence of the diabetes. Some mice showing long-term remission of type 1 diabetes after anti-TCRβ therapy underwent i.p. injections of anti-PDL1 at 140–170 days after the onset of type 1 diabetes as previously described [27]. Anti-PDL1 was dosed at 25 mg/kg on day 0, 12.5 mg/kg on day 2 and 12.5 mg/kg on day 4. Blood glucose levels were monitored for recurrence of diabetes.
Intraperitoneal GTT
Non-diabetic NOD mice, diabetic isotype-treated NOD mice, NOD mice treated with anti-TCRβ for the prevention of type 1 diabetes (11 weeks post-treatment) and NOD mice treated with anti-TCRβ to induce a remission of type 1 diabetes (11 weeks post-treatment) were fasted for 5 h. After fasting, 1 g/kg glucose was given by i.p. injection and the blood glucose level was measured at 0, 15, 30, 45, 60 and 120 min after injection.
Serum insulin measurements
Blood was harvested at random from NOD-scid mice, non-diabetic NOD mice, diabetic isotype-treated NOD mice and anti-TCRβ-treated NOD mice in remission (10 and 60 days post-treatment). Following isolation of the serum, the insulin concentration was measured using the Mercodia Mouse Insulin ELISA kit (Uppsala, Sweden).
Islet histology and scoring
Pancreatic tissue was harvested from NOD-scid mice, non-diabetic NOD mice, diabetic isotype-treated NOD mice and anti-TCRβ-treated NOD mice in remission (10 and 60 days post-treatment). The pancreatic tissue samples were fixed in formalin, embedded in paraffin and sectioned at three non-overlapping levels. The sections were stained with haematoxylin and eosin (H&E) for a histological analysis of insulitis as previously described [28]. The islets were scored individually as follows: 0, no lesions; 1, peri-insulitis; 2, <50% islet destruction; and 3, >50% islet destruction.
Skin transplantation
Skin transplants were performed as previously described [29] at 12–18 weeks after the onset of type 1 diabetes in anti-TCRβ-treated NOD mice. Ear skin (1.0 cm2) from WT BALB/c donors was grafted onto the flanks of NOD recipients. The grafts were covered with sterile bandages, which were removed 7 days post-transplant. Thereafter, the grafts were monitored daily for rejection (>80% necrosis).
Flow cytometry analyses
Spleen, pancreatic draining lymph nodes (PLNs), pancreatic infiltrating cells and peripheral blood mononuclear cells (PBMCs) were harvested from non-diabetic NOD mice, diabetic isotype-treated NOD mice and anti-TCRβ-treated NOD mice in remission (10 and 60 days after the last dose). Cell suspensions were prepared and stained with fluorescence-conjugated antibodies as indicated. Flow cytometry was performed using a BD FACSCalibur machine (BD Biosciences, San Jose, CA, USA).
Statistical analyses
Statistical analyses of curves of the percentage of diabetes-free mice were performed using the logrank test in GraphPad Prism (GraphPad Software, San Diego, CA, USA). Immune cell subset analyses and serum insulin levels were compared using the Kruskal–Wallis test with a Dunn’s multiple comparison post-test to determine statistical significance. All other statistical analyses were performed using an unpaired, two-tailed Student’s t test to determine statistical significance at the levels indicated.
Results
Transient anti-TCRβ treatment prevents type 1 diabetes with minimal inflammatory cytokine production
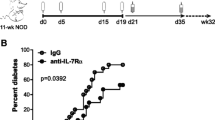
Due to our previous observations of the strong effects of anti-TCRβ on antigen-specific T cell responses both in vitro and in vivo, the antibody was tested for its ability to inhibit T cell-mediated autoimmunity in an acute model of type 1 diabetes. RIP-OVAhi mice adoptively transferred with OT-I, OT-II and OVA323–339 peptide-pulsed DCs were treated with either anti-TCRβ or PBS. All the mice treated with anti-TCRβ remained diabetes-free long after the adoptive transfer, whereas all the PBS-treated mice developed diabetes within 11 days (Fig. 1a, b).

Transient anti-TCRβ therapy prevents type 1 diabetes. (a, b) Diabetes-free survival and glucose measurements in RIP-OVAhi mice (n = 4 per group) injected with purified OT-I, OT-II and OVA-pulsed DCs and given a single dose of anti-TCRβ (solid lines) or PBS (dashed lines). *p < 0.05. (c) Diabetes-free survival in 8-week-old normoglycaemic NOD mice treated with anti-TCRβ (solid line, n = 10), isotype (dotted line, n = 10) or anti-CD3 (dashed line, n = 8). *p < 0.05 vs isotype. Individual glucose measurements are shown for (d) anti-TCRβ (e) isotype and (f) anti-CD3
Anti-TCRβ was next tested in a more clinically relevant model of type 1 diabetes. Eight-week-old female NOD mice were treated with anti-TCRβ, anti-CD3 or isotype and monitored until 40 weeks of age. Whereas the anti-CD3 and isotype treatments were ineffective, the anti-TCRβ treatment prevented disease in 90% of these mice (Fig. 1c–f). These experiments demonstrate the efficacy of anti-TCRβ treatment in preventing both the acute induction of type 1 diabetes in RIP-OVAhi mice and the spontaneous development of type 1 diabetes in NOD mice.
Since other antibodies targeting T cell signalling components have shown severe side effects such as cytokine release syndrome [30], cytokine production was measured following the anti-TCRβ, anti-CD3 and isotype treatments. Serum levels of IL-2, IFN-γ, TNF-α and IL-6 were consistently low or undetectable in the isotype control group. In contrast, 1 mg/kg anti-CD3 significantly elevated the serum levels of IL-2 and TNF-α at both 1.5 h and 4 h, and of IFN-γ at 12 h, compared with anti-TCRβ (Fig. 2a–d). Increasing doses of anti-TCRβ elicited significantly lower levels of serum IL-2, IL-6 and IFN-γ and undetectable levels of TNF-α when compared with anti-CD3 (Fig. 2e–h). Normoglycaemic NOD mice exhibited significantly lower levels of IL-2, IL-6 and IFN-γ when injected with anti-TCRβ compared with anti-CD3. Similarly, NOD mice with new-onset diabetes that were injected with anti-TCRβ had significantly lower levels of IL-2 and IL-6 production compared with those injected with anti-CD3 (Fig. 2i–l). Thus, anti-TCRβ has the potential to prevent T cell-mediated autoimmunity in both RIP-OVAhi and NOD mice, with minimal inflammatory cytokine release.

Inflammatory cytokine production after anti-TCRβ treatment. (a–d) WT C57BL/6 mice were injected with anti-CD3 (black bars), anti-TCRβ (white bars) or isotype (grey bars), and serum levels of IL-2, IFN-γ, TNF-α and IL-6 were measured by ELISA at the indicated times after injection. (e–h) WT C57BL/6 mice were injected with anti-CD3, anti-TCRβ or isotype at the indicated concentrations. Serum IL-2, TNF-α and IL-6 were measured at 4 h and IFN-γ at 12 h after injection. (i–l) Non-diabetic and diabetic NOD mice were injected with 1 mg/kg of anti-CD3, anti-TCRβ or isotype. Serum IL-2, TNF-α and IL-6 were measured at 4 h and IFN-γ at 12 h after injection. Experiments were performed in duplicate with each bar representing measurements from three mice; the representative results of one experiment are presented. *p < 0.05. ND, not detectable
Anti-TCRβ therapy restores durable self-tolerance in new-onset diabetic NOD mice
The NOD mice with new-onset diabetes were treated with anti-TCRβ, anti-CD3 or isotype. Similarly to anti-CD3, anti-TCRβ was able to reverse the type 1 diabetes in 73% of the female NOD mice (Fig. 3a–d). The durability of this self-tolerance was tested by challenging anti-TCRβ-treated NOD mice (long after remission) with an MHC-mismatched BALB/c skin allograft (Fig. 3e–g). Although all the recipients acutely rejected the skin allografts with a mean graft survival time of 10 ± 1 days (Fig. 3f), they remained diabetes free even several weeks after graft rejection (Fig. 3g). Together, these results demonstrate that brief treatment with anti-TCRβ after the onset of type 1 diabetes can reverse the disease in a majority of NOD mice. This restoration of self-tolerance is maintained long after stopping treatment and does not interfere with the ability of the immune system to produce a robust response against foreign alloantigens.

Transient therapy with anti-TCRβ restores durable self-tolerance in NOD mice with new-onset diabetes. (a) Diabetes-free survival in new-onset diabetic NOD mice treated with anti-TCRβ (solid line, n = 15), anti-CD3 (dashed line, n = 5) or isotype (dotted line, n = 12). *p < 0.05 vs isotype. Individual glucose measurements are shown for (b) anti-TCRβ (c) isotype and (d) anti-CD3. (e–g) Individual glucose measurements and graft survival in NOD mice with new-onset diabetes treated with anti-TCRβ and then transplanted with BALB/c skin allografts at 12–18 weeks after the onset of diabetes (n = 3)
Anti-TCRβ-treated NOD mice have preserved pancreatic islet function with reduced inflammatory infiltration
Anti-TCRβ-treated and healthy non-diabetic NOD mice each displayed normal reactions to an i.p. GTT (Fig. 4a). In addition, anti-TCRβ-treated mice had significantly higher serum insulin levels at day 10 and day 60 after treatment than isotype-treated diabetic mice (Fig. 4b). The serum insulin levels in the anti-TCRβ-treated groups were similar to those obtained from normoglycaemic NOD-scid and non-diabetic NOD mice. Histological analysis and pathological scoring revealed a large degree of infiltration and destruction of islets in the isotype-treated diabetic mice and reduced infiltration and preserved islet architecture in the anti-TCRβ-treated cohorts (Fig. 4c–h). Thus, early-onset diabetic NOD mice treated with anti-TCRβ maintain normal glucose tolerance and insulin levels and have decreased islet destruction compared with isotype-treated mice.

Anti-TCRβ-treated NOD mice respond normally to glucose challenge and have reduced insulitis. (a) Blood glucose measurements over time in diabetic (squares), non-diabetic (circles), anti-TCRβ pre-onset treated (diamonds) and anti-TCRβ post-onset treated (triangles) NOD mice after i.p. glucose challenge. Each point represents the mean ± SD of values from three mice. *p < 0.05 vs diabetic mice. (b) Serum insulin levels in NOD-scid (n = 4), non-diabetic (n = 6), diabetic (n = 6) and anti-TCRβ treated NOD mice at 10 (n = 6) and 60 (n = 6) days after treatment measured by ELISA. The bars represent mean ± SD. *p < 0.05. Representative islets from H&E-stained sections of pancreatic tissue from (c) NOD-scid, (d) non-diabetic, (e) diabetic and anti-TCRβ treated NOD mice at (f) 10 and (g) 60 days after treatment. (h) Islets were scored 0 (white), 1 (light grey), 2 (dark grey) and 3 (black) as described in the “Methods”. The percentage of islets within each category is depicted in the bar graph. Each bar represents a total of 60 islets (20 from each of three mice)
Anti-TCRβ therapy reduces gut-homing receptor CCR9+ T cells without causing broad lymphocyte depletion in NOD mice
To understand the effects of anti-TCRβ on the immune cell populations in NOD mice, the percentages of CD4+ and CD8+ in the total lymphocyte populations were measured in spleen, PLNs, pancreatic tissue and PBMCs by flow cytometry (see electronic supplementary material [ESM] Fig. 1 for dot plots). There were no significant differences in the frequency of CD4+ (Fig. 5a–c) or CD8+ (Fig. 5e–g) T cells in the spleen, PLNs or pancreatic tissue. In addition, there were no significant differences in the percentage of Tregs within the CD4+ T cell population in any of the tissues tested (data not shown). The CD4+ and CD8+ T cell populations in PBMCs were reduced in the anti-TCRβ-treated day 10 group compared with the diabetic group (Fig. 5d, h). These findings indicate that anti-TCRβ treatment causes limited lymphocyte depletion in NOD mice. The expression of the gut-homing receptor CCR9 on CD4+ T cells showed little difference in the tissues tested (Fig. 5i–l). Further analysis revealed that anti-TCRβ-treated day 10 mice had significant reductions in the percentage of CCR9+CD8+ T cells in all the lymphoid and pancreatic tissues tested compared with diabetic mice (Fig. 5m–p and ESM Fig. 2). This reduction in CCR9+CD8+ T cells compared with diabetic controls was also seen in the PLNs and spleen of anti-TCRβ treated mice at day 60. Thus, transient anti-TCRβ therapy in NOD mice led to reductions in the percentage of gut-homing CCR9+CD8+ T cells.

Anti-TCRβ affects gut-homing receptor-expressing T cells. The tissues indicated were harvested from non-diabetic (white), diabetic (black) and anti-TCR treated NOD mice at 10 (light grey) and 60 (dark grey) days after treatment for flow cytometry analysis of the lymphocyte populations. The bar graphs show the percentage of (a–h) CD4+ and CD8+ cells, (i–l) CCR9+ cells within CD4+ cells, and (m–p) CCR9+ cells within CD8+ cells. The sample size is shown below each bar, representing the mean ± SD. *p < 0.05
Anti-TCRβ therapy increases PD-1 expression on T cells that maintains self-tolerance in NOD mice
Further analysis of the T cells in anti-TCRβ-treated NOD mice showed a significant increase in the expression of the co-inhibitory molecule PD-1 on the surface of the CD4+ T cells in the spleen at day 10 and in PBMCs at days 10 and 60 after treatment (Fig. 6a–d and ESM Fig. 3). The expression of PD-1 on the surface of the CD8+ T cells was also significantly increased in the pancreatic tissue of the anti-TCRβ-treated NOD mice at day 10 and in the PBMCs of the anti-TCRβ-treated NOD mice at day 60 (Fig. 6e–h).

PD-1 expression on T cells increases after anti-TCRβ treatment. The tissues indicated were harvested from non-diabetic (white), diabetic (black) and anti-TCRβ treated NOD mice at 10 (light grey) and 60 (dark grey) days after treatment for flow cytometry analysis of the lymphocyte populations. The bar graphs show the mean fluorescence intensity (MFI) of PD-1 expression on (a–d) CD4+ and (e–h) CD8+ T cell populations in each tissue. The sample size is shown below each bar, representing the mean ± SD. *p < 0.05
PD-1 signalling in activated T cells provides an inhibitory signal to the T cells when it binds to its ligand PDL1, which is constitutively expressed on the surface of many cell types including cells in the pancreatic islets [31]. To determine whether signalling through PD-1 was important for the restoration of self-tolerance by anti-TCRβ therapy in NOD mice, anti-TCRβ-treated NOD mice that had achieved a long-term remission of type 1 diabetes (Fig. 7a) were treated with a brief course of anti-PDL1 mAb. Within 5 days of the initial administration of anti-PDL1, all four NOD mice developed diabetes (Fig. 7b, c). This rapid recurrence of diabetes was not due to a pancreatic beta cell toxicity of anti-PDL1, as administration to healthy WT C57BL/6 mice did not induce hyperglycaemia. This evidence demonstrates the critical role that PD-1/PDL1 signalling plays in maintaining anti-TCRβ-induced self-tolerance in NOD mice.

Maintenance of self-tolerance restored by anti-TCRβ treatment requires signals through the PD-1 signalling pathway. NOD mice with new-onset diabetes (solid lines, n = 4) that were treated with anti-TCRβ (as in Fig. 3a) were injected with three doses of anti-PDL1 at 140–170 days after the onset of the diabetes. Normoglycaemic WT C57BL/6 control mice (dash-dot lines, n = 4) received an identical anti-PDL1 regimen. (a) Blood glucose levels in the NOD mice with new-onset diabetes treated with anti-TCRβ demonstrating a remission of the diabetes. (b) Diabetes-free survival and (c) blood glucose levels following anti-PDL1 administration
Discussion
Over a hundred different strategies have been reported for preventing type 1 diabetes in NOD mice [6], but only a few treatments reliably reverse type 1 diabetes after its onset in NOD mice [32]. Notably, anti-CD3 therapy can reverse [10] but not prevent [33] type 1 diabetes in NOD mice unless it is given during the neonatal time period [34]. In contrast, anti-TCRβ treatment is effective both before and after the onset of type 1 diabetes in NOD mice. The reason for this difference in the ability to prevent diabetes in NOD mice is unclear but may be related to differences in the half-lives of the antibodies, as anti-TCR has a longer half-life [35], or in the ability of the antibodies to stimulate T cells to proliferate and produce cytokines [36]. Although anti-TCRβ treatment has previously been found to be effective in preventing type 1 diabetes in NOD mice [26], this is the first demonstration of a long-term reversal of disease using this TCRβ-targeting antibody.
There are a number of instances in which T cell-targeted antibody therapies have been shown to initiate a cytokine release syndrome, limiting their use in treating allograft rejection or autoimmunity [30]. However, in this study, anti-TCRβ elicited little inflammatory cytokine release compared with anti-CD3 in this and other publications, even at high doses [37]. Clinical studies have supported this unique property of TCR-targeted antibody therapies, as anti-human TCRα mAb (T10B9) used as an induction therapy in transplantation had fewer side-effects related to cytokine release compared with anti-CD3 mAb (OKT3) [22]. This may help to translate anti-TCR mAb back into clinical use for autoimmune diseases such as type 1 diabetes as it could allow for an expansion of the dose and timing of treatment beyond that of CD3-targeted regimens [17, 19, 21].
Previous work has demonstrated the effectiveness of anti-TCRβ therapy, both alone and in combination with other treatments, in abrogating alloimmune T cell responses in order to prolong allograft survival in mice [24, 25]. Unlike the effects in C57BL/6 allograft models, in which anti-TCRβ caused a broad depletion of CD4+ and CD8+ T cells [25], treatment with anti-TCRβ in NOD mice led to only limited lymphocyte depletion in PBMCs without enhancement of the Treg population. These differences in lymphocyte depletion may represent other interstrain differences between C57BL/6 and NOD mice, as has been reported for other antibody-induced effects [38]. The activation status of the immune system in NOD mice is different from that in C57BL/6 mice, and it has been observed that T cells in adult NOD mice are resistant to activation-induced cell death by agonistic antibodies such as anti-CD3 [39]. This lack of broad lymphocyte depletion may explain how anti-TCRβ-treated NOD mice could still mount an effective immune response to reject BALB/c skin allografts. Importantly, this inflammatory event did not result in a recurrence of diabetes, suggesting the restoration of a durable self-tolerance by anti-TCRβ.
Moreover, a significant reduction in the population of CCR9+CD8+ T cells in all tissues suggests that anti-TCRβ engagement leads to a decreased expression of the gut-homing CCR9 receptor and therefore less infiltration of gut-associated tissues by these cells. A population of CCR9+ T cells that produce the cytokine IL-21 was recently reported to be enriched in the pancreas and PLNs of diabetic NOD mice. This subset of T helper cells is necessary for development of type 1 diabetes through direct effects on cytotoxic T cells [40]. It may thus be possible that anti-TCRβ exerts its protective effects by preventing help being given by CCR9+ T cells to the cytotoxic T cells that play a critical role in pathogenesis of type 1 diabetes [41].
New-onset diabetic NOD mice treated with anti-TCRβ also showed an increased expression of the co-inhibitory molecule PD-1 on CD4+ T cells in the spleen and PBMCs and on CD8+ T cells in PLNs at day 10 after treatment. These values remained high in both CD4+ and CD8+ T cells in PBMCs at day 60. In addition, signalling through the PD-1/PDL1 pathway was critical for the maintenance of anti-TCRβ-induced self-tolerance, as demonstrated by the PDL1-blocking antibody experiments. These results are in line with previous data describing the key role of PD-1 in maintaining self-tolerance to autoantigens by suppressing infiltration into the pancreas [42]. The PD-1/PDL1 pathway has also been implicated in the reversal of type 1 diabetes by several other therapies such as antigen-specific insulin and anti-CD3 by inducing anergy of the pathogenic T cells [27, 43]. PD-1 signalling is also known to drive the exhaustion of activated T cells in chronic infections and tumours [44]. However, the role of T cell exhaustion in models of type 1 diabetes remains unclear.
Overall, our studies show a safe and effective method for reversing the spontaneous onset of type 1 diabetes in NOD mice using transient anti-TCRβ therapy. The therapy induced durable self-tolerance without a broad depletion of T cells and without affecting their ability to respond to foreign antigens. Long-term remission from type 1 diabetes correlated with reduced CCR9+ T cells counts and increased PD-1 expression, which was critical for the maintenance of self-tolerance. These results have important implications for the future clinical use of TCR-targeted immune therapies for type 1 diabetes.
Abbreviations
- CCR9:
-
Chemokine receptor 9
- DC:
-
Dendritic cell
- H&E:
-
Haematoxylin and eosin
- mAb:
-
Monoclonal antibody
- NOD-scid :
-
NOD.Cg-Prkdc scidIl2rgtm1Wjl/SzJ
- OVA:
-
Ovalbumin
- PBMC:
-
Peripheral blood mononuclear cell
- PD-1:
-
Programmed death 1
- PDL1:
-
Programmed death ligand 1
- PLN:
-
Pancreatic draining lymph node
- Rag1 −/−OT-I:
-
B6.129S7-Rag1 tm1MomTg(TcraTcrb)1100Mjb
- Rag1 −/−OT-II:
-
B6.129S7-Rag1 tm1MomTg(TcraTcrb)425Cbn
- RIP-OVAhi :
-
C57BL/6-Tg(Ins2-OVA)59Wehi/WehiJ
- TCRβ:
-
T cell receptor β-chain
- Treg:
-
Regulatory T cell
- WT:
-
Wild-type
References
Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G, Group ES (2009) Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet 373:2027–2033
van Belle TL, Coppieters KT, von Herrath MG (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91:79–118
Roep BO (2003) The role of T cells in the pathogenesis of type 1 diabetes: from cause to cure. Diabetologia 46:305–321
Anderson MS, Bluestone JA (2005) The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 23:447–485
Pagliuca FW, Millman JR, Gurtler M et al (2014) Generation of functional human pancreatic beta cells in vitro. Cell 159:428–439
Atkinson MA, Leiter EH (1999) The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med 5:601–604
Herold KC, Vignali DA, Cooke A, Bluestone JA (2013) Type 1 diabetes: translating mechanistic observations into effective clinical outcomes. Nat Rev Immunol 13:243–256
Tian J, Clare-Salzler M, Herschenfeld A et al (1996) Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat Med 2:1348–1353
Grinberg-Bleyer Y, Baeyens A, You S et al (2010) IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 207:1871–1878
Chatenoud L, Thervet E, Primo J, Bach JF (1994) Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A 91:123–127
Chen W, Xie A, Chan L (2013) Mechanistic basis of immunotherapies for type 1 diabetes mellitus. Transl Res 161:217–229
Ludvigsson J, Krisky D, Casas R et al (2012) GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med 366:433–442
Wherrett DK, Bundy B, Becker DJ et al (2011) Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet 378:319–327
Orban T, Bundy B, Becker DJ et al (2011) Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet 378:412–419
Krause S, Landherr U, Agardh CD et al (2014) GAD autoantibody affinity in adult patients with latent autoimmune diabetes, the study participants of a GAD65 vaccination trial. Diabetes Care 37:1675–1680
Long SA, Rieck M, Sanda S et al (2012) Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes 61:2340–2348
Sherry N, Hagopian W, Ludvigsson J et al (2011) Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 378:487–497
Aronson R, Gottlieb PA, Christiansen JS et al (2014) Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 37:2746–2754
Ambery P, Donner TW, Biswas N, Donaldson J, Parkin J, Dayan CM (2014) Efficacy and safety of low-dose otelixizumab anti-CD3 monoclonal antibody in preserving C-peptide secretion in adolescent type 1 diabetes: DEFEND-2, a randomized, placebo-controlled, double-blind, multi-centre study. Diabet Med 31:399–402
Staeva TP, Chatenoud L, Insel R, Atkinson MA (2013) Recent lessons learned from prevention and recent-onset type 1 diabetes immunotherapy trials. Diabetes 62:9–17
Demeester S, Keymeulen B, Kaufman L et al (2015) Preexisting insulin autoantibodies predict efficacy of otelixizumab in preserving residual beta-cell function in recent-onset type 1 diabetes. Diabetes Care. doi:10.2337/dc14-1575
Waid TH, Lucas BA, Thompson JS et al (1997) Treatment of renal allograft rejection with T10B9.1A31 or OKT3: final analysis of a phase II clinical trial. Transplantation 64:274–281
Knight RJ, Kurrle R, McClain J et al (1994) Clinical evaluation of induction immunosuppression with a murine IgG2b monoclonal antibody (BMA 031) directed toward the human alpha/beta-T cell receptor. Transplantation 57:1581–1588
Schroder PM, Khattar M, Deng R, Xie A, Chen W, Stepkowski SM (2013) Transient combination therapy targeting the immune synapse abrogates T cell responses and prolongs allograft survival in mice. PLoS One 8:e69397
Miyahara Y, Khattar M, Schroder PM et al (2012) Anti-TCRbeta mAb induces long-term allograft survival by reducing antigen-reactive T cells and sparing regulatory T cells. Am J Transplant 12:1409–1418
Sempe P, Bedossa P, Richard MF, Villa MC, Bach JF, Boitard C (1991) Anti-alpha/beta T cell receptor monoclonal antibody provides an efficient therapy for autoimmune diabetes in nonobese diabetic (NOD) mice. Eur J Immunol 21:1163–1169
Fife BT, Guleria I, Gubbels Bupp M et al (2006) Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med 203:2737–2747
Ablamunits V, Henegariu O, Hansen JB et al (2012) Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes 61:145–154
Garrod KR, Cahalan MD (2008) Murine skin transplantation. J Vis Exp 634
Bugelski PJ, Achuthanandam R, Capocasale RJ, Treacy G, Bouman-Thio E (2009) Monoclonal antibody-induced cytokine-release syndrome. Exp Rev Clin Immunol 5:499–521
Keir ME, Liang SC, Guleria I et al (2006) Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 203:883–895
Roep BO, Atkinson M, von Herrath M (2004) Satisfaction (not) guaranteed: re-evaluating the use of animal models of type 1 diabetes. Nat Rev Immunol 4:989–997
Chatenoud L, Primo J, Bach JF (1997) CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 158:2947–2954
Hayward AR, Shreiber M (1989) Neonatal injection of CD3 antibody into nonobese diabetic mice reduces the incidence of insulitis and diabetes. J Immunol 143:1555–1559
Henrickson M, Reid J, Bellet JS, Sawchuk SS, Hirsch R (1995) Comparison of in vivo efficacy and mechanism of action of antimurine monoclonal antibodies directed against TCR alpha beta (H57-597) and CD3 (145-2C11). Transplantation 60:828–835
Kubo RT, Born W, Kappler JW, Marrack P, Pigeon M (1989) Characterization of a monoclonal antibody which detects all murine alpha beta T cell receptors. J Immunol 142:2736–2742
Ferran C, Sheehan K, Dy M et al (1990) Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: further evidence for transient in vivo T cell activation. Eur J Immunol 20:509–515
Ferran C, Dy M, Sheehan K et al (1991) Inter-mouse strain differences in the in vivo anti-CD3 induced cytokine release. Clin Exp Immunol 86:537–543
Yang W, Hussain S, Mi QS, Santamaria P, Delovitch TL (2004) Perturbed homeostasis of peripheral T cells elicits decreased susceptibility to anti-CD3-induced apoptosis in prediabetic nonobese diabetic mice. J Immunol 173:4407–4416
McGuire HM, Vogelzang A, Ma CS et al (2011) A subset of interleukin-21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 34:602–615
Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG (2009) Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 155:173–181
Pauken KE, Jenkins MK, Azuma M, Fife BT (2013) PD-1, but not PD-L1, expressed by islet-reactive CD4+ T cells suppresses infiltration of the pancreas during type 1 diabetes. Diabetes 62:2859–2869
Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T (2005) Establishment of NOD-Pdcd1 –/– mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A 102:11823–11828
Wherry EJ (2011) T cell exhaustion. Nat Immunol 12:492–499
Acknowledgements
We would like to acknowledge B. Chojnacki, manager of the flow cytometry core facility at the University of Toledo College of Medicine, for his assistance with the acquisition of some of the flow cytometry data for this paper. We would also like to acknowledge K. Goans, associate director of the Department of Laboratory Animal Resources at the University of Toledo College of Medicine, for her assistance in the housing and care of the animals used for this study.
Funding
This work was supported by funds from the National Institutes of Health (grant number 5RO1AI090244 to SMS) and the UT-Biomedical Science Innovation Award (to SMS). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
MK, PMS, YM, WC and SMS contributed to the conception and design of the study. MK, PMS, CEB, YM, RV, SM and BM were involved in the acquisition, analysis and interpretation of the data. MK, PMS, YM, and SMS drafted the article. MK, PMS, CEB, WC, RV, SM and BM were involved in revising the article for important intellectual content. All authors have approved the final version of this manuscript for publication. SMS is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Additional information
Paul M. Schroder and Mithun Khattar contributed equally to this work and should be considered joint first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 440 kb)
ESM Fig. 2
(PDF 388 kb)
ESM Fig. 3
(PDF 238 kb)
Rights and permissions
About this article

Cite this article
Schroder, P.M., Khattar, M., Baum, C.E. et al. PD-1-dependent restoration of self-tolerance in the NOD mouse model of diabetes after transient anti-TCRβ mAb therapy. Diabetologia 58, 1309–1318 (2015). https://doi.org/10.1007/s00125-015-3564-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3564-1