
Abstract
Aim/hypothesis
Type 2 diabetes is a complex disease characterised by hyperglycaemia, hyperinsulinaemia, dyslipidaemia and insulin resistance accompanied by inflammation. Previously, we showed that mice lacking the Wdr13 gene had increased islet mass due to enhanced beta cell proliferation. We hypothesised that introgression of a Wdr13-null mutation, a beta cell-proliferative phenotype, into Lepr db/db mice, a beta cell-destructive phenotype, might rescue the diabetic phenotype of the latter.
Methods
Wdr13-deficient mice were crossed with Lepr db/db mice to generate mice with the double mutation. We measured various serum metabolic variables of Wdr13 +/0 Lepr db/db and Wdr13 −/0 Lepr db/db mice. Further, we analysed the histopathology and gene expression of peroxisome proliferator-activated receptor (PPAR)γ and, activator protein (AP)1 targets in various metabolic tissues.
Results
Lepr db/db mice with the Wdr13 deletion had a massively increased islet mass, hyperinsulinaemia and adipocyte hypertrophy. The increase in beta cell mass in Wdr13 −/0 Lepr db/db mice was due to an increase in beta cell proliferation. Hypertrophy of adipocytes may be the result of increase in transcription of Pparg and its target genes, leading in turn to increased expression of several lipogenic genes. We also observed a significant decrease in the expression of AP1 and nuclear factor κ light chain enhancer of activated B cells (NFκB) target genes involved in inflammation.
Conclusions/interpretation
This study provides evidence that loss of WD repeat domain 13 (WDR13) protein in the Lepr db/db mouse model of diabetes is beneficial. Based on these findings, we suggest that WDR13 may be a potential drug target for ameliorating hyperglycaemia and inflammation in diabetic conditions.
Similar content being viewed by others

Introduction
Type 2 diabetes is a worldwide disease, estimated to affect more than 340 million people, with approximately 3.4 million associated deaths per year [1]. Type 2 diabetes is associated with increases in obesity, insulin resistance, inflammation and pancreatic beta cell malfunction [2, 3]. Insulin resistance is one of the main reasons for beta cell failure in animal models of type 2 diabetes and in humans [4]. The increase in obesity and the resulting insulin resistance initially trigger beta cell expansion. However, long-term insulin resistance leads to beta cell stress and, ultimately, beta cell failure [5]. Beta cell mass and function are also regulated by a variety of adipokines, namely leptin, adiponectin, resistin and visfatin secreted from adipose tissue [6].
The leptin receptor mutant (Lepr db/db) mouse is a model of type 2 diabetes. The mutant mice show hyperphagia due to defects in leptin signalling, which leads to obesity, hyperglycaemia, hyperinsulinaemia, dyslipidaemia, insulin resistance and inflammation at around 3 months [7]. Glucotoxicity and lipotoxicity are two secondary phenomena implicated in the pathology of type 2 diabetes, resulting from excessive glucose and/or lipids. Glucotoxicity occurs via dysregulation of genes involved in gluconeogenesis, glycogenolysis and glycolysis: glucose 6-phosphate, phosphoenolpyruvate carboxykinase (PEPCK), glycogen phosphorylase and glucokinase in liver [8, 9]. Lipotoxicity occurs via impaired hepatic fatty acid metabolism and adipose tissue dysfunction [10]. Obesity is accompanied by a chronic low-grade inflammation in adipose tissue and liver characterised by infiltration of cytokines, chemokines and macrophages together with activation of pro-inflammatory transcription factors such as activator protein (AP)1, nuclear factor κ light chain enhancer of activated B cells (NFκB) and 5′ UTR regulatory element (PU.1) [11, 12].
WDR13 is a member of the WD repeats protein family and occurs in most tissues, being abundant in the pancreas, brain, testis and ovary [13, 14]. Wdr13-knockout mice have increased pancreatic beta cell proliferation, which leads to increased islet mass, hyperinsulinaemia and better glucose clearance, notwithstanding mild obesity at a later age [15]. We hypothesised that introgression of a Wdr13-null mutation, a beta cell-proliferative phenotype, into Lepr db/db mice, a beta cell-destructive phenotype, might rescue the diabetic phenotype of the latter. Here, we show that the deletion of Wdr13 gene in Lepr db/db mice significantly reduces circulating blood glucose and inflammation.
Methods
Generation of mice and metabolic measurements
The Institutional Animal Ethics Committee of the Centre for Cellular and Molecular Biology (CCMB) approved all animal experiments. To generate congenic Wdr13-knockout C57BL/6J mice (JAX mice, Bar Harbor, ME, USA), CD1 Wdr13-knockout mice were backcrossed for nine generations. To generate double heterozygotes for Wdr13 and Lepr mutant alleles, Wdr13-heterozygous females were mated with heterozygous males of the B6.BKS-Lepr db strain (JAX mice, Bar Harbor, ME, USA [stock 000697]). Finally, double-heterozygous male and female mice obtained from this cross were bred to generate mice with the double mutation in the Wdr13 and Lepr genes. Animals were genotyped using PCR. Body weight, food intake, glucose and insulin tolerance testing and insulin measurements were performed as described previously [15]. Circulating triacylglycerol was measured using the glycerol-3-phosphate oxidase/phenol + aminophenazone (GPO/PAP) method (Coral Clinical Systems, Goa, India). For triacylglycerol content, frozen liver (~50 mg) was homogenised in 500 μl PBS and chloroform-extracted lipids were dried using a Speedvac and re-dissolved in isopropanol. Triacylglycerol was quantified in 96 well plates. NEFA were measured using a half micro test kit (Roche, Basel, Switzerland).
Histology western blot analysis and real-time PCR
Histology and western blot analysis were performed as described [15]. All the tissues for histology, western blot and real-time PCR analyses were collected from mice fasted for 16 h. Haematoxylin and eosin (H&E) staining and immunostaining were carried out on 4 μm sections, mounted on positively charged slides, from tissues fixed overnight in 4% paraformaldehyde and embedded in paraffin. Immunostaining was performed using antibodies to jun proto-oncogene (c-Jun) ([sc-45] Santa Cruz Biotechnology, Paso Robles, CA, USA), Ki-67 ([AB9260] EMD Millipore, Billerica, MA, USA) and Mac-2 (also known as galectin-3) ([CL8942AP] Cedarlane Labs, ON, Canada). Antibodies to c-Jun and Ki-67 were detected using a biotin–streptavidin–horseradish peroxidase (HRP) system and Mac-2 was detected using an HRP-conjugated antibody and developed with diaminobenzidine (DAB) substrate ([11718096001] Roche). Positive cells were counted manually using Axioskop (AxioVision software, www.zeiss.co.in/microscopy/en_in/downloads/axiovision.html). A minimum of 1,000 beta cells was considered for the quantification of each group. For Oil Red O (Sigma-Aldrich, Bangalore, India) staining, liver tissues were frozen in OCT compound (Sigma-Aldrich) and 8 μm sections were mounted on slides and stained with 0.5% Oil Red O solution. Western blots were performed using antibodies to nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor, alpha (IκBα) ([4812S] Cell Signaling Technology, Beverly, MA, USA), c-Jun, p-c-Jun ([sc-822] Santa Cruz Biotechnology) and β-actin ([ab8226] Abcam, Tokyo, Japan). Total RNA was extracted from frozen tissues using an RNeasy mini kit (Qiagen, Limburg, the Netherlands). Reverse transcription was performed using ImProm II Reverse Transcriptase kit (Promega, Madison, WI, USA). Quantitative (q)RT-PCR was performed using Ex Taq Sybr Green (Takara, New Delhi, India). Primer sequences are presented in electronic supplementary material (ESM) Table 1.
Apoptotic assay
Tissue sections were prepared as described previously [15]. TUNEL assays were performed using the DeadEnd Fluorometric TUNEL System (Promega). Confocal images were recorded and TUNEL-positive beta cells were counted manually from a total of 5–6 mice per genotype.
Pparg promoter reporter assay
The Pparg promoter was cloned in pGL3 (Promega) according to Zhu et al [16].
Statistical analysis
The unpaired Student’s t test was used, with significance at p < 0.05. Data are represented as mean ± SEM.
Results
Wdr13−/0Leprdb/db mice resist weight loss
We measured the body weights of Wdr13 −/0 Lepr db/db and Lepr db/db mice to 6 months of age. Wdr13 −/0 Lepr db/db mice weighed less than Lepr db/db mice at 3 months (Fig. 1a). In Lepr db/db mice there was no difference in body weight at 3 and 6 months whereas Wdr13 −/0 Lepr db/db showed a steady increase in body weight throughout the experiment to 6 months (Fig. 1b). This difference between these two genotypes was attributable to a significant loss in body weight in Lepr db/db mice after 4 months of age owing to diabetic complications, whereas Wdr13 −/0 Lepr db/db mice showed significant body weight gain from 4 to 6 months (Fig. 1c). No significant difference in food intake was observed between these two genotypes (Fig. 1d).

The effect of Wdr13 gene deletion on body weight and food intake in Lepr db/db mice. (a, b) Body weight measurements taken at (a) 3 and (b) 6 months. (c) The weight gain was calculated as the difference in body weight at 4 and 6 months (d) Feed consumption of various genotypes. n = 6–8 mice per group. Dark grey bars, Wdr13 +/0 Lepr +/+ mice; light grey bars, Wdr13 −/0 Lepr +/+ mice; black bars, Wdr13 +/0 Lepr db/db mice; white bars, Wdr13 −/0 Lepr db/db mice. *p < 0.05
Wdr13−/0Leprdb/db mice showed improved metabolic phenotype and better glucose tolerance
To understand the effect of Wdr13 deletion, we measured glucose, insulin, triacylglycerol and NEFA in Wdr13 −/0 Lepr db/db and Lepr db/db mice and the respective control genotypes. Fasting and fed blood glucose levels were significantly lower in Wdr13 −/0 Lepr db/db mice than in Lepr db/db mice at both 3 and 6 months (Fig. 2a–d). Interestingly, at 3 months, the insulin levels were comparable in these two genotypes (Fig. 2e, g). Lepr db/db mice were already diabetic whereas Wdr13 −/0 Lepr db/db mice had a significantly lower glucose level than Lepr db/db mice. As expected, the insulin level in Lepr db/db mice at 6 months was lower than that at 3 months because of the destruction of beta cells, whereas in the Wdr13 −/0 Lepr db/db mice insulin level continued to increase (Fig. 2e–h) and that was reflected in the glucose values. In addition to reduced blood glucose, serum triacylglycerol was also significantly lower in Wdr13 −/0 Lepr db/db mice at 6 months (Fig. 2j). Similarly, NEFA was significantly lower in Wdr13 −/0 Lepr db/db than in Lepr db/db mice at this age (Fig. 2l).

The effect of Wdr13 gene deletion on metabolic variables in Lepr db/db mice at 3 and 6 months. Blood glucose levels after 16 h of fasting (a, b) and in the random fed state (c, d). Serum insulin levels after 16 h of fasting (e, f) and in the random fed state (g, h). (i, j) Serum triacylglycerol after 16 h of fasting. (k, l) Serum NEFA after 16 h of fasting. n = 5–8 mice per group. Dark grey bars, Wdr13 +/0 Lepr +/+ mice; light grey bars, Wdr13 −/0 Lepr +/+ mice; black bars, Wdr13 +/0 Lepr db/db mice; white bars, Wdr13 −/0 Lepr db/db mice. *p < 0.05 and **p < 0.01
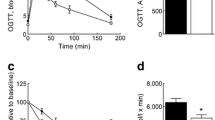
The glucose tolerance test (GTT) showed no significant difference in glucose clearance between Wdr13 −/0 Lepr db/db and Lepr db/db mice at 3 months (Fig. 3a) except for a significantly low basal glucose level in Wdr13 −/0 Lepr db/db mice as already described above (Fig. 2a). At 6 months, Wdr13 −/0 Lepr db/db mice cleared glucose efficiently, restoring to basal values after 120 min, while Lepr db/db mice showed severely impaired glucose tolerance as reflected by glucose levels of >55.5 mmol/l, even at 120 min (Fig. 3b). Insulin tolerance test at 3 months showed better insulin sensitivity in Wdr13 −/0 Lepr db/db mice compared with Lepr db/db mice, the former showing compromised insulin sensitivity when compared with wild type (Fig. 3c). At 6 months we observed insulin resistance in Wdr13 −/0 Lepr db/db mice even though the basal glucose level was significantly lower than in Lepr db/db mice (Fig. 3d).

The effect of Wdr13 gene deletion on glucose clearance and insulin sensitivity in Lepr db/db mice. GTT at (a) 3 and (b) 6 months. ITT at (c) 3 and (d) 6 months. n = 5–8 mice per group. Circles, Wdr13 +/0 Lepr +/+ mice; squares, Wdr13 −/0 Lepr +/+ mice; triangles, Wdr13 +/0 Lepr db/db mice; inverted triangles, Wdr13 −/0 Lepr db/db mice. *p < 0.05 and **p < 0.01
Wdr13−/0Leprdb/db mice have greater islet area
To understand the effect of Wdr13 gene deletion in Lepr db/db mice, we examined the pancreatic histology. At 3 months there was no difference in islet area between Lepr db/db and Wdr13 −/0 Lepr db/db mice (ESM Fig. 1c, d) whereas it was significantly higher in the Wdr13 −/0 Lepr db/db mice at 6 months (Fig. 4a). Moreover, the islet boundaries were well defined and the islets appeared healthier in the Wdr13 −/0 Lepr db/db mice. Thus, improved glucose clearance in Wdr13 −/0 Lepr db/db mice might be due to the increase in the islet area.

The effect of Wdr13 deletion on the histology of pancreas, liver and adipose tissue in Lepr db/db mice at 6 months. (a) H&E staining of the pancreas with quantification of islet area. Scale bars, 100 μm. (b) Immunostaining for Ki-67-positive cells (arrowheads). Scale bars, 50 μm. (c) Liver H&E and Oil Red O staining. Scale bars, 10 μm and 20 μm (n = 5 mice per group). (d) H&E staining of the epididymal fat pad. Scale bars, 100 μm. Dark grey bars, Wdr13 +/0 Lepr +/+ mice; light grey bars, Wdr13 −/0 Lepr +/+ mice; black bars, Wdr13 +/0 Lepr db/db mice; white bars, Wdr13 −/0 Lepr db/db mice. *p < 0.05
Wdr13−/0Leprdb/db mice have increased beta cell proliferation
We assayed beta cell proliferation in Wdr13 −/0 Lepr db/db mice using Ki-67 antibody. The number of Ki-67-positive cells was twofold higher (p = 0.07) in Wdr13 −/0 Lepr db/db mice than in Lepr db/db mice at 6 months (Fig. 4b). Therefore, the increase in islet area may be attributed to the increase in beta cell proliferation in Wdr13 −/0 Lepr db/db. We did not observe any difference in the number of apoptotic beta cells using TUNEL assay in the genotypes at 3 months (ESM Fig. 1a, b). It was difficult to find any apoptotic cells at 6 months in Lepr db/db mice as there were only disintegrated and damaged islets and it was not possible to compare the extent of apoptosis.
Wdr13−/0Leprdb/db mice showed reduced triacylglycerol in liver and hypertrophy of adipocytes
Liver histology at 6 months showed a drastic reduction in lipid droplets in Wdr13 −/0 Lepr db/db mice compared with Lepr db/db mice (Fig. 4c). Analysis of liver triacylglycerol content further confirmed the reduced lipid accumulation in Wdr13 −/0 Lepr db/db mice (Fig. 4c). Histological examination of epididymal fat pad sections revealed hypertrophy of adipocytes in both the Lepr db/db and Wdr13 −/0 Lepr db/db mice (Fig. 4d). However, Wdr13 −/0 Lepr db/db mice showed more hypertrophy with compact and regular adipocyte morphology, in contrast to Lepr db/db mice.
Wdr13−/0Leprdb/db mice showed reduced pancreatic inflammation
WDR13 acts as a transcriptional activator of c-Jun in the presence of high c-Jun N-terminal kinase (JNK) activity (V. P. Singh, S. Katta, S. Kumar, unpublished data) and c-Jun regulates many AP1 and NFκB target genes. JNK signalling promotes insulin resistance in beta cells [17]. c-Jun immunostaining in beta cells showed reduced levels in Wdr13 −/0 Lepr db/db mice (Fig. 5a). In addition, analysis of inflammatory markers such as Emr1 (macrophage) and Mcp-1 (also known as Ccl2) in total pancreatic extracts showed reduced expression in Wdr13 −/0 Lepr db/db mice (Fig. 5b).

The effect of Wdr13 deletion on pancreatic inflammation. (a) c-Jun immunostaining of the pancreas. Scale bars, 50 μm. (b) mRNA levels of genes involved in inflammation. Expression has been calculated relative to Wdr13 +/0 Lepr db/db mice, set at 1. Black bars, Wdr13 +/0 Lepr db/db mice; white bars, Wdr13 −/0 Lepr db/db mice. *p < 0.05
Improved adipose tissue function in Wdr13−/0Leprdb/db mice
Adipose tissue gene expression analysis showed a consistent increase in Acc1 (also known as Acaca), Acc2 (also known as Acacb), Fasn, Srebp1c (also known as Srebf1) and Scd1 in Wdr13 −/0 Lepr db/db mice, indicating increased lipogenesis and improved fat-accumulation capacity of adipocytes (Fig. 6a). As PPARγ is the master regulator of adipose tissue function, and adipose tissue functions are correlated mainly with its transcription factors, we examined the expression of Pparg and its target genes. A significant—approximately 20-fold—increase in Pparg mRNA level was observed in Wdr13 −/0 mice compared with wild type. Similarly, Wdr13 −/0 Lepr db/db mice showed twofold increase in Pparg expression in contrast to Lepr db/db (Fig. 6b). As expected, there was also an increase in the expression of its target genes, Ap2 (also known as Fabp4), Glut4 and Lpl (Fig. 6c). In Wdr13 −/0 Lepr db/db mice, we observed a fourfold increase in mRNA levels of Pepck-c (also known as Pck1) compared with Lepr db/db (Fig. 6d), consistent with a reduction in circulating NEFA (Fig. 2l), as reported earlier, which suggests increased de novo glyceroneogenesis.

The effect of Wdr13 deletion on adipose tissue lipogenesis and inflammation in Lepr db/db mice at 6 months. (a) mRNA levels of genes involved in lipid metabolism. (b) qRT-PCR for Pparg. (c) mRNA levels of PPARγ target genes. (d) Pepck mRNA levels in adipose tissue. (e) Western blot for phosphorylated and total c-Jun levels. (f) mRNA levels of AP1 and NFκB target genes. (g) mRNA levels of genes involved in inflammation. (h) Western blot for IκBα in adipose tissue. Β-actin was used as loading control. (i) Mac-2 staining for macrophages. Ip10 is also known as Cxcl10; Mpa-2l is also known as Gbp6. In (c), (d) and (f) expression has been calculated relative to Wdr13 +/0 Lepr db/db mice, set at 1. Scale bars, 20 μm. Dark grey bars, Wdr13 +/0 Lepr +/+ mice; light grey bars, Wdr13 −/0 Lepr +/+ mice; black bars, Wdr13 +/0 Lepr db/db mice; white bars, Wdr13 −/0 Lepr db/db mice. n = 6 mice per group. *p < 0.05
Wdr13−/0Leprdb/db mice showed reduced inflammation in adipose tissue
As mentioned above, Wdr13 acts as a transcriptional activator of c-Jun and it is known that Lepr db/db mice have high JNK activity and high p-c-Jun level, which activate AP1-specific pro-inflammatory target genes. Western blot data showed similar levels of p-c-Jun in Lepr db/db and Wdr13 −/0 Lepr db/db mice (Fig. 6e). Thus, we analysed the expression of inflammatory genes in the Wdr13 −/0 Lepr db/db mice. In adipose tissue, in spite of increased hypertrophy in Wdr13 −/0 Lepr db/db mice, a decrease in inflammatory markers such as Emr1, Socs3 and the transcription factor Pu.1 along with AP1 and NFκB target genes containing AP1 sites was observed compared with Lepr db/db (Fig. 6f, g). As Wdr13 −/0 Lepr db/db mice showed decreased expression of NFκB target genes we analysed the protein levels of IκBα. In Wdr13 −/0 Leprdb/db mice IκBα levels were significantly higher than in Lepr db/db, implying a reduction in inflammation through retention of NFκB by IκBα in the cytoplasm as NFκB–IκBα complex (Fig. 6h). Mac-2 staining for the accumulation of macrophages around adipose tissue displayed less crown-like structure (CLS) formation in Wdr13 −/0 Leprdb/db mice (Fig. 6i).
Next, we studied the expression of genes involved in the major pathways of glucose and fatty acid metabolism in liver. Wdr13 −/0 Lepr db/db mice showed a significant decrease in the expression of key gluconeogenic enzyme pyruvate dehydrogenase kinase, isozyme 4 (PDK4) and lipogenic enzymes Fas (TNF receptor superfamily member 6) (FAS) and stearoyl-coenzyme A desaturase 1 (SCD1), whereas increased expression of acetyl-coenzyme A carboxylase α (ACC) and diacylglycerol O-acyltransferase 2 (DGAT2) was observed (ESM Fig. 2b). The liver inflammatory genes Emr1 and Socs3 were also downregulated in the Wdr13 −/0 Lepr db/db mice compared with Lepr db/db (ESM Fig. 2b). Furthermore, the presence of intact IκBα protein (ESM Fig. 2a) and reduced protein levels of p-c-Jun (ESM Fig. 2c) were consistent with the reduced expression of inflammatory genes in the livers of Wdr13 −/0 Lepr db/db mice.
Discussion
We show that deletion of Wdr13 in Lepr db/db mice significantly reduces levels of circulating blood glucose and NEFA. We observed that, compared with Lepr db/db, Wdr13 −/0 Lepr db/db mice had higher beta cell proliferation, massively increased islet mass, and hyperinsulinaemia, accompanied by better glucose clearance. Wdr13 −/0 Lepr db/db mice displayed improved adipose tissue function via increased PPARγ and expression of its downstream target genes. These mice were protected from obesity-induced inflammation by reduced expression of AP1- and NFκB-induced target genes.
Lepr db/db mice gain weight rapidly till about 3 months, and then progressively lose body weight because of severe impairment of glucose and lipid metabolism [7]. In the absence of Wdr13, Lepr db/db mice had lower body weight at 3 months (Fig. 1a). The lower body weight of the Wdr13 −/0 Lepr db/db mice might have been a reflection of a lower birthweight of Wdr13-knockout mice [15], or Wdr13 deletion per se might have prevented body weight gain in the Wdr13 −/0 Lepr db/db mice from birth to 3 months. Beyond 3 months, the Wdr13 −/0 Lepr db/db mice continued to increase in body weight till the termination of the experiment at 6 months, in contrast to Lepr db/db mice. These data were consistent with the significantly improved glucose homeostasis observed in the absence of Wdr13 in Lepr db/db mice.
Diabetes in Lepr db/db mice is characterised by hyperinsulinaemia, hyperglycaemia, obesity and insulin resistance [7]. At 3 months Wdr13 −/0 Lepr db/db and Lepr db/db mice showed similar insulin levels (Fig. 2e, g). Interestingly, glucose levels were significantly reduced in the Wdr13 −/0 Lepr db/db mice, indicating better insulin sensitivity in the absence of Wdr13 in Lepr db/db, which was further confirmed by insulin tolerance test (ITT) at 3 months (Fig. 3c). However, Wdr13 −/0 Lepr db/db mice were still hyperglycaemic and insulin resistant when compared with wild type (Figs 2a, c and 3c). Further, at 6 months Lepr db/db mice showed severe hyperglycaemia along with a reduction in insulin levels, as expected, because of beta cell destruction [4]. However, insulin levels were higher in the Wdr13 −/0 Lepr db/db mice. The effect of this increase in insulin level was reflected in better glucose clearance in Wdr13 −/0 Lepr db/db mice than in Lepr db/db mice. At 6 months Wdr13 −/0 Lepr db/db mice did not respond to exogenous insulin in contrast to Lepr db/db mice (Fig. 3d). This difference might reflect the very high levels of endogenous insulin levels intrinsic to Wdr13 −/0 Lepr db/db mice. As ITT makes a limited contribution to an understanding of insulin resistance in a tissue-specific manner, the possibility remains that insulin resistance may vary in different tissues and adipose tissue insulin sensitivity in Wdr13 −/0 Lepr db/db mice might have improved. A continuous demand for insulin in Lepr db/db mice leads to pancreatic beta cell death [4]. As there is no change in the number of apoptotic beta cells in Wdr13 −/0 Lepr db/db mice compared with Lepr db/db (ESM Fig. 1a) and WDR13 is a negative regulator of pancreatic beta cell proliferation [15], the hyperinsulinaemia in the Wdr13 −/0 Lepr db/db mice is primarily due to the increased islet mass resulting from the pancreatic beta cell proliferation, as shown by Ki-67 staining (Fig. 4a, b). These results are consistent with our previous studies on the role of Wdr13 in beta cell proliferation [15].
In liver, insulin suppresses gluconeogenesis and increases lipogenesis [8, 18], whereas in adipose tissue it promotes glucose and fatty acid uptake [19], and suppresses lipolysis [20]. PPARγ plays a crucial role in the control of glucose and lipid homeostasis [21–26]. We found that the Wdr13 −/0 Lepr db/db mice had a significant increase in mRNA levels of Pparg and its downstream target genes, Glut4 (also known as Slc2a4), Lpl and Ap2 (also known as Fabp4) (Fig. 6b, c). The Wdr13 −/0 Lepr db/db mice showed increased expression of lipogenic genes such as Acc1, Acc2, Fasn, Scd1 and Srebp1c in adipose tissue in comparison with Lepr db/db mice, indicating increased lipogenesis (Fig. 6a). These data are consistent with the adipose tissue hypertrophy seen in Wdr13 −/0 Lepr db/db mice (Fig. 4d). It appears that the increased lipogenesis may be one of the contributing factors responsible for lowering the glucose levels, notwithstanding higher insulin levels, for the maintenance of glucose homeostasis. Wdr13 is expressed in adipocytes and our data support the cell-autonomous role of Wdr13 in adipocytes as shown by enhanced adipocyte lipogenesis in the absence of Wdr13. It is very likely that the increased insulin levels in Wdr13-knockout animals would reduce lipolysis in adipose tissue, resulting in increased fat storage. To delineate the relative contribution of these two possible mechanisms for an increase in fat accumulation in adipocytes, one would need a tissue-specific knockout of Wdr13 gene. Previous studies have shown that genetic deletion of adipose tissue chaperones increased de novo lipogenesis in adipose tissue, rendering the mice resistant to metabolic syndrome [27–30]. The enhanced lipogenesis in adipose tissue of Wdr13 −/0 Lepr db/db mice was complemented by a reduction in lipolysis, as shown by lower levels of triacylglycerol and NEFA in serum compared with Leprdb/db [31, 32]. In the livers of Wdr13 −/0 Lepr db/db mice, the picture from Oil Red O staining and triacylglycerol content was clearly of a reduction in lipogenesis (Fig. 4c). To understand this reduction, we analysed the mRNA levels of six lipogenic genes and that of the transcription factor sterol regulatory element binding transcription factor 1 (SREBP1C). With the exception of Acc1 and Acc2, the genes showed downregulation. It is possible that downregulation of FAS, which catalyses the downstream step of Acc1/Acc2 in fatty acid synthesis, may contribute to the reduction in de novo lipogenesis [33]. The upregulation of Acc1 and Acc2 observed by us at the mRNA level is an apparent contradiction of the Wdr13 −/0 Lepr db/db phenotype. It is known that the activities of these two enzymes are not only regulated at the transcription level but also through post-translational modification, specifically phosphorylation [34]. Further investigations are needed to resolve this.
Type 2 diabetes and obesity have been associated with chronic low-grade inflammation [11, 35]. Consistent with the improved functionality of adipose tissue and other metabolic variables in the Wdr13 −/0 Lepr db/db mice, a significant reduction was observed in inflammatory markers (Fig. 6g) and macrophage infiltration (Fig. 6i) in these mice. Similarly, inflammation was reduced in the pancreas and liver of Wdr13 −/0 Lepr db/db mice (Fig. 5 and ESM Fig. 2b), demonstrating a systemic effect due to reduced adipose tissue inflammation. The intact IκBα in the Wdr13 −/0 Lepr db/db mice further strengthens our proposition that inflammation is reduced in various metabolic tissues of these mice (Fig. 6h and ESM Fig. 2a). A parallel study showed that Wdr13 interacts with c-Jun and acts as a transcriptional co-activator in the presence of high JNK activity in various cell types (V. P. Singh, S. Katta, S. Kumar, unpublished data). It is established that Lepr db/db mice and other mouse models of diabetes exhibit high JNK activity due to inflammation. Analysis of AP1 and NFκB target genes containing AP1 sites showed reduced expression in Wdr13 −/0 Lepr db/db mice (Figs 5b and 6f and ESM Fig. 2), strengthening the above findings. Wdr13 is expressed in various metabolic tissues: pancreatic islets, liver, adipose and muscle [15]. It is possible that the reduced expression of AP1 target genes (pro-inflammatory) in these tissues may also contribute to the beneficial effect of loss of WDR13. The Pparg promoter contains an AP1 site and WDR13 binds with c-Jun. Promoter analysis of Pparg2 in 3T3L1 cells showed a significant increase after c-Jun overexpression (ESM Fig. 3), suggesting an interesting and indirect role of Wdr13 in Pparg2 regulation. However, at present we do not understand the regulation of Pparg2 promoter by WDR13 in the presence of high JNK activity.
In conclusion, we examined in detail the phenotype of pancreas, liver and adipose tissue using whole-body Wdr13 knockout and provide evidence of a beneficial effect of loss of this protein in Lepr db/db mice. Interestingly, our data suggest that inflammation is reduced in the pancreas, liver and adipose tissue as a result of the downregulation of AP1-target genes in these tissues. Given the expression of Wdr13 in multiple tissues including brain, tissue-specific knockout of this gene will further help in explaining the phenotype we describe.
Abbreviations
- AP:
-
Activator protein
- CCMB:
-
Centre for Cellular and Molecular Biology
- GTT:
-
Glucose tolerance test
- H&E:
-
Haematoxylin and eosin
- HRP:
-
Horseradish peroxidase
- IκBα:
-
Nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor α
- ITT:
-
Insulin tolerance test
- JNK:
-
c-Jun N-terminal kinase
- NFκB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PPAR:
-
Peroxisome proliferator-activated receptor
- qRT-PCR:
-
Quantitative RT-PCR
- WDR13:
-
WD repeat domain 13
References
Roglic G, Unwin N, Bennett PH et al (2005) The burden of mortality attributable to diabetes: realistic estimates for the year 2000. Diabetes Care 28:2130–2135
Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625
Wajchenberg BL (2007) Beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev 28:187–218
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Invest 116:1802–1812
Robertson R, Zhou H, Zhang T, Harmon JS (2007) Chronic oxidative stress as a mechanism for glucose toxicity of the beta cell in type 2 diabetes. Cell Biochem Biophys 48:139–146
Dunmore SJ, Brown JE (2013) The role of adipokines in beta-cell failure of type 2 diabetes. J Endocrinol 216:T37–T45
Sharma K, McCue P, Dunn SR (2003) Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol 284:F1138–F1144
Postic C, Dentin R, Girard J (2004) Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab 30:398–408
Matschinsky FM, Magnuson MA, Zelent D et al (2006) The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes 55:1–12
Lewis GF, Carpentier A, Adeli K, Giacca A (2002) Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 23:201–229
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29:415–445
Singh BN, Suresh A, UmaPrasad G et al (2003) A highly conserved human gene encoding a novel member of WD-repeat family of proteins (WDR13). Genomics 81:315–328
Suresh A, Shah V, Rani DS et al (2005) A mouse gene encoding a novel member of the WD family of proteins is highly conserved and predominantly expressed in the testis (Wdr13). Mol Reprod Dev 72:299–310
Singh VP, Lakshmi BJ, Singh S et al (2012) Lack of Wdr13 gene in mice leads to enhanced pancreatic beta cell proliferation, hyperinsulinemia and mild obesity. PLoS One 7:e38685
Zhu Y, Qi C, Korenberg JR et al (1995) Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc Natl Acad Sci U S A 92:7921–7925
Lanuza-Masdeu J, Arevalo MI, Vila C, Barbera A, Gomis R, Caelles C (2013) In vivo JNK activation in pancreatic beta-cells leads to glucose intolerance caused by insulin resistance in pancreas. Diabetes 62:2308–2317
Muoio DM, Newgard CB (2008) Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9:193–205
Girard J, Perdereau D, Foufelle F, Prip-Buus C, Ferre P (1994) Regulation of lipogenic enzyme gene expression by nutrients and hormones. FASEB J 8:36–42
Bergman RN, Ader M (2000) Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab 11:351–356
Medina-Gomez G, Gray SL, Yetukuri L et al (2007) PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 3:e64
Miles PD, Barak Y, He W, Evans RM, Olefsky JM (2000) Improved insulin-sensitivity in mice heterozygous for PPAR-gamma deficiency. J Clin Invest 105:287–292
Kubota N, Terauchi Y, Miki H et al (1999) PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell 4:597–609
He W, Barak Y, Hevener A et al (2003) Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A 100:15712–15717
Kim HS, Hwang YC, Koo SH et al (2013) PPAR-gamma activation increases insulin secretion through the up-regulation of the free fatty acid receptor GPR40 in pancreatic beta-cells. PLoS One 8:e50128
Ferre P (2004) The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes 53(Suppl 1):S43–S50
Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS (2008) Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 134:933–944
Kuriyama H, Liang G, Engelking LJ, Horton JD, Goldstein JL, Brown MS (2005) Compensatory increase in fatty acid synthesis in adipose tissue of mice with conditional deficiency of SCAP in liver. Cell Metab 1:41–51
Waki H, Park KW, Mitro N et al (2007) The small molecule harmine is an antidiabetic cell-type-specific regulator of PPARgamma expression. Cell Metab 5:357–370
Maeda K, Cao H, Kono K et al (2005) Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab 1:107–119
Magkos F, Fabbrini E, Conte C, Patterson BW, Klein S (2012) Relationship between adipose tissue lipolytic activity and skeletal muscle insulin resistance in nondiabetic women. J Clin Endocrinol Metab 97:E1219–E1223
Mittelman SD, Bergman RN (2000) Inhibition of lipolysis causes suppression of endogenous glucose production independent of changes in insulin. Am J Physiol Endocrinol Metab 279:E630–E637
Strable MS, Ntambi JM (2010) Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit Rev Biochem Mol Biol 45:199–214
Fullerton MD, Galic S, Marcinko K et al (2013) Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19:1649–1654
Galic S, Fullerton MD, Schertzer JD et al (2011) Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest 121:4903–4915
Acknowledgements
We thank A. Raj (CCMB, Hyderabad, India) for his technical assistance in histology and N. Rangaraj (CCMB, Hyderabad, India) for assistance in microscopy. We thank A. B. Siva for critical reading of the manuscript and suggestions, and L. Singh for support.
Funding
The Department of Biotechnology (DBT), Government of India, New Delhi and the Council of Scientific and Industrial Research (CSIR), New Delhi supported this research.
Contribution statement
All authors were responsible for the conception and design, or analysis and interpretation of data; and drafting the article or revising it critically for important intellectual content. All authors gave final approval of the version to be published. Satish Kumar is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Duality of interest
The authors confirm that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Vijay P. Singh and Chandrashekaran Gurunathan contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
(PDF 11,540 kb)
ESM Fig. 2
(PDF 2,899 kb)
ESM Fig. 3
(PDF 1,354 kb)
ESM Table 1
(PDF 70 kb)
Rights and permissions
About this article

Cite this article
Singh, V.P., Gurunathan, C., Singh, S. et al. Genetic deletion of Wdr13 improves the metabolic phenotype of Lepr db/db mice by modulating AP1 and PPARγ target genes. Diabetologia 58, 384–392 (2015). https://doi.org/10.1007/s00125-014-3438-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3438-y