
Abstract
Aims/hypothesis
We evaluated the effects of a combination triple antioxidant therapy on measures of cardiovascular autonomic neuropathy (CAN) and myocardial blood flow (MBF) in patients with type 1 diabetes.
Methods
This was a randomised, parallel, placebo-controlled trial. Participants were allocated to interventions by sequentially numbered, opaque, sealed envelopes provided to the research pharmacist. All participants and examiners were masked to treatment allocation. Participants were evaluated by cardiovascular autonomic reflex testing, positron emission tomography with [11C]meta-hydroxyephedrine ([11C]HED) and [13N]ammonia, and adenosine stress testing. Markers of oxidative stress included 24 h urinary F2-isoprostanes. Diabetic peripheral neuropathy (DPN) was evaluated by symptoms, signs, electrophysiology and intra-epidermal nerve fibre density. Randomised participants included 44 eligible adults with type 1 diabetes and mild-to-moderate CAN, who were aged 46 ± 11 years and had HbA1c 58 ± 5 mmol/mol (7.5 ± 1.0%), with no evidence of ischaemic heart disease. Participants underwent a 24-month intervention, consisting of antioxidant treatment with allopurinol, α-lipoic acid and nicotinamide, or placebo. The main outcome was change in the global [11C]HED retention index (RI) at 24 months in participants on the active drug compared with those on placebo.
Results
We analysed data from 44 participants (22 per group). After adjusting for age, sex and in-trial HbA1c, the antioxidant regimen was associated with a slight, but significant worsening of the global [11C]HED left ventricle RI (−0.010 [95% CI −0.020, −0.001] p = 0.045) compared with placebo. There were no significant differences at follow-up between antioxidant treatment and placebo in the global MBF, coronary flow reserve, or in measures of DPN and markers of oxidative stress. The majority of adverse events were of mild-to-moderate severity and did not differ between groups
Conclusions/interpretation
In this cohort of type 1 diabetes patients with mild-to-moderate CAN, a combination antioxidant treatment regimen did not prevent progression of CAN, had no beneficial effects on myocardial perfusion or DPN, and may have been detrimental. However, a larger study is necessary to assess the underlying causes of these findings.
Trial registration
ClinicalTrials.gov NCT00116207
Funding
The study was funded by the JDRF Centre for the Study of Complications in Diabetes; and by grants P60DK020572 and P30DK092926 to the Michigan Diabetes Research and Training Centre (MDRTC) and the Michigan Center for Diabetes Translational Research (MCDTR) from the National Institute of Diabetes and Digestive and Kidney Diseases.
Similar content being viewed by others


Introduction
Death from cardiovascular disease (CVD) remains the main cause of excess mortality rates in patients with type 1 diabetes [1]. Cardiovascular autonomic neuropathy (CAN) is associated with silent myocardial ischaemia and predicts enhanced cardiac risk. The development of CAN is a function of complex interactions between degrees of glycaemic control, disease duration, age-related neuronal attrition, and systolic and diastolic blood pressure [2]. These promote progressive autonomic neural dysfunction in a fashion that parallels the development of peripheral neuropathy, e.g. beginning distally and progressing proximally. Our data [3] and those of others [4] have confirmed that there is a compensatory increase in cardiac sympathetic tone in response to subclinical peripheral denervation early in the progression of CAN in patients with type 1 diabetes. Sympathetic denervation follows later, beginning at the apex of the ventricles, and progresses towards the base.
Oxidative stress is implicated in the development and progression of the chronic complications of diabetes, including CAN [3, 5–8]. We have previously reported that indices of oxidative stress were correlated with cardiac sympathetic dysinnervation and impaired myocardial vascular responsiveness in type 1 diabetes patients [3].
The goal of the current study was to understand the interrelationships between left ventricle (LV) sympathetic dysinnervation, altered myocardial blood flow (MBF) regulation, impaired neurotrophism and oxidative stress in type 1 diabetes. We postulated that attenuation of oxidative stress would result in the prevention or reversal of CAN. Phase II randomised controlled trials using the antioxidant α-lipoic acid (ALA) had previously shown some favourable effects on indices of heart rate variability [9]. However, the reported effects of various individual antioxidants in preventing or reversing diabetic peripheral neuropathy (DPN) or CAN have been inconsistent and disappointing [2, 6, 7]. We postulated that the failure of single antioxidant agents reflected a failure to address the multiple pathways that generate oxidative stress and are thought to promote complications of diabetes.
In the current study, we used a combination antioxidant therapeutic regimen aimed to address three different steps in the generation of oxidative damage: (1) xanthine-oxidase inhibition with allopurinol (to prevent oxygen free radical formation); (2) scavenging of oxygen free radicals with ALA; and (3) poly (ADP-ribose) synthase inhibition with nicotinamide (to reduce the downstream consequences of oxidative stress). We hypothesised that if several pathways were targeted at the same time with a combined antioxidant regimen, this would more effectively attenuate oxidative stress than single agents, and prevent deficits in LV sympathetic innervation and deficits in myocardial vascular responsiveness in participants with type 1 diabetes and mild-to-moderate CAN. We also conducted a comprehensive analysis of the effects of this antioxidant cocktail on measures of DPN in these patients.
Methods
Study design and patient population
This trial was a prospective, randomised, double-blind, placebo-controlled parallel group study. Participants with type 1 diabetes with early complications were randomly assigned to a combination antioxidant regimen or to placebo. Inclusion criteria required a diagnosis of type 1 diabetes (WHO), age 18 to 65 years, HbA1c <75 mmol/mol (9%), stable diabetes control over the previous 3 months, the presence of mild non-proliferative diabetic retinopathy (at least level ≥20 in the Early Treatment of Diabetic Retinopathy Scale) [10] or of microalbuminuria, and the presence of CAN, defined as a distal defect in [11C]meta-hydroxyephedrine ([11C]HED) retention involving at least 10% of the LV. These criteria were selected on the basis of previously obtained evidence describing mild deficits in LV [11C]HED retention in participants with mild non-proliferative retinopathy or mild albuminuria in spite of normal cardiovascular reflex testing [3]. Participants with any of the following were excluded: pre-existent CVD (coronary artery disease, positive stress test, congestive heart failure, arrhythmias, ventricular structural abnormalities, valvular disease), uncontrolled hypertension, severe systemic or inflammatory diseases (which could be associated with an increased likelihood of retinopathy, nephropathy or neuropathy), previous kidney, pancreas or cardiac transplantation, pregnancy or lactation in women, history of drug or alcohol dependence, and use of any other medications known to interfere with the uptake or metabolism of catecholamines (including immunosuppressants, tricyclic antidepressants, monoamine oxidase inhibitors and cocaine).
Intervention
Allopurinol (300 mg daily), ALA (600 mg twice daily) and nicotinamide (750 mg twice daily), or matched oral placebos were administered for 24 months. The administration of each individual active drug or placebo component was titrated in consecutive weeks (first ALA, then nicotinamide, finally allopurinol) such that the participant began receiving full therapeutic doses of all the medications 3 weeks post-randomisation. Participants were allocated to interventions by sequentially numbered, opaque, sealed envelopes provided to the research pharmacist. All investigators, clinic staff and participants were blinded to treatment group assignment.
Participants were evaluated at 3 month intervals and the following recorded: data on vital signs, results of physical examination, HbA1c values, compliance with study drugs, laboratory safety and adverse events. Follow-up evaluations were obtained immediately after completion of the 24 month treatment. All patients were required to stay on the study drug until the day of final evaluations.
All participants signed a written informed consent document and the University of Michigan Institutional Review Board approved the study.
CAN evaluations
Patients were evaluated for the presence of CAN using positron emission tomography (PET) imaging of the LV with [11C]HED and cardiovascular autonomic reflex tests at baseline and follow-up.
Because autonomic function may be altered by various factors, all participants were required to fast and avoid caffeine, tobacco products, and prescription and over-the-counter medicines (except usual insulin regimen) for at least 8 h before CAN testing. Participants who experienced hypoglycaemia (defined as blood glucose ≤2.775 mmol/l and/or signs/symptoms of hypoglycaemia) after midnight prior to the above examinations, as well as those with acute illness in the previous 48 h were rescheduled.
Evaluation of LV sympathetic innervation by PET imaging with [11C]HED
All PET studies were performed on an ECAT ExactHR+PET scanner (Siemens Medical Solutions USA, Knoxville, TN, USA)). Participants were positioned in the PET scanner gantry and 74 MBq [13N]ammonia was injected i.v., followed by a 2 min brief scan to visualise the heart. This ‘scout’ [13N]ammonia scan was used to adjust the bed position so that the heart was at the centre of the scanner’s field of view and to confirm lack of resting perfusion abnormalities in the participant’s LV. After 30 min, 740 MBq [11C]HED was injected i.v. while a 40 min dynamic PET data acquisition sequence was started (22 image frames; 12 × 10 s, 2 × 30 s, 2 × 60 s, 2 × 150 s, 2 × 300 s, 2 × 600 s) as described [11, 12].
[ 11 C]HED retention analysis
The LV wall in the eight short axis slices from the [11C]HED study (encompassing the LV from apex to base) was divided into 60 sectors to generate 480 independent LV regions. The [11C]HED radioactivity concentration measured in each sector in the final image frame (30–40 min) was normalised to the calculated integral of the total radioactivity in the blood pool throughout the PET study, and the [11C]HED retention index (RI) (in [ml blood] min−1 [ml tissue]−1) was obtained for each LV sector, as previously described [11]. Polar maps of regional [11C]HED retention were generated and visually inspected for [11C]HED retention deficits. A quantitative measure of the degree of cardiac denervation in each participant was generated by statistically comparing the [11C]HED RI value of each sector in the participant’s [11C]HED polar map with the mean and SD of the RI data for that sector in our database of healthy non-diabetic participants (age range 20 to 78 years, n = 15 men, n = 18 women, n = 33 total). Using this standard z score analysis [12], sectors in the participant’s [11C]HED polar map with RI values more than 2.5 SD below the healthy control mean value were considered to be regions with ‘abnormal’ [11C]HED retention.
Cardiovascular autonomic reflex tests
The standardised cardiovascular autonomic reflex tests (CARTs) included the paced R-R response to deep breathing, the Valsalva manoeuvre and postural changes in blood pressure as described [13], and were performed with Viking Quest II (Nicolet, Middleton, WI, USA).
Evaluation of MBF and coronary flow reserve
Evaluation of dynamic MBF and coronary flow reserve (CFR), a measure of endothelial function, was done at baseline and follow-up, using [13N]ammonia at rest and during pharmacologically induced (i.v. adenosine) coronary vasodilation as previously described [14].
DPN evaluations
DPN evaluations comprised the following: (1) assessment of symptoms using the Michigan Neuropathy Screening Instrument (MNSI), a validated neuropathy questionnaire [15]; (2) comprehensive neurological evaluation performed by board-certified neurologists; (3) nerve conduction studies (NCS) including the sural, peroneal and median nerves with standardisation for limb temperature as described [16]; (4) quantitative sensory testing (QST) for cold detection and vibration perception thresholds; and (5) quantitative sudomotor axon reflex testing (QSART). In addition, skin biopsies were obtained to assess intra-epidermal nerve fibre density (IENFD), a measure of small-fibre neuropathy, to capture earliest changes associated with DPN and to evaluate the effects of intervention. All these tests were done at baseline and follow-up. Skin biopsies were performed as described [17]. Briefly 3 mm skin samples were obtained from the ankle and proximal thigh on the non-dominant side after intradermal local anaesthesia with 1% (wt/vol.) lidocaine. The tissue was fixed for 6 h in paraformaldehyde lysine phosphate before transfer to cryoprotectant and cutting into 50 μm sections using a freezing sliding microtome (Leica CM1850, Leica Microsystems, Buffalo Grove, IL, USA). Sections were stained with the pan axonal marker PGP 9.5 (Serotec, Raleigh, NC, USA)
Epidermal nerve fibres were counted using established criteria as described [17]. The final IENFD measurement was derived by taking the mean of four to six randomly selected individual sections.
Evaluation of systemic oxidative stress
Systemic oxidative stress was evaluated by measuring free F2-isoprostanes, a reliable biomarker for assessment of oxidative stress and lipid peroxidation in vivo, from 24 h urine sample collections, and quantified using gas chromatography–mass spectrometry as described by Liu et al [18]. Deuterated internal standard was added to the free F2-isoprostanes and solid-phase extraction was completed. A moiety of pentafluorobenzyl was then introduced to the molecule and the hydroxyl groups were capped by trimethylsiyl derivatisation. A selective-ion monitoring technique was used to analyse the derivatives of F2-isoprostanes and the internal standard; the ions monitored were m/z 569 and m/z 573, respectively.
Outcome measures
The primary outcome was the change in the global [11C]HED RI at 24 months in participants taking the active drug compared with those on placebo. Three secondary endpoints were also specified: endothelial dysfunction as measured by the global CFR, systemic oxidative stress as assessed by 24 h urinary free F2-isoprostanes and inflammation as assessed by high-sensitivity C-reactive protein (CRP). All other outcomes were specified as exploratory; these included changes in: (1) regional [11C]HED RI and regional CFR; (2) resting MBF; (3) cardiovascular autonomic reflex testing; (4) measures of DPN, defined either as clinically confirmed peripheral neuropathy requiring two positive responses among neuropathic symptoms (pain, numbness, paraesthesia); and sensory signs and abnormal ankle reflexes, confirmed by NCS abnormalities involving two or more of the sural, peroneal and median nerves [16]; and (5) changes in IENFD.
Statistical analysis
Randomisation
Block randomisation was based on a block size of 4, with stratification by sex and age, where age was divided into two strata: ≤45 vs >45 years old.
Sample size
The SD of [11C]HED was assumed to be 0.0113, based on our previously published data of the magnitude of the RI deficit observed in the distal myocardial segments of patients with moderate to severe CAN [19]. With 15 participants per group, we estimated 80% power to detect a change of 0.015 between the mean [11C]HED RI of the placebo-treated and actively treated groups, when testing the null hypothesis using a two-tailed two-sample t test at a 5% level of significance. A 20% loss due to early withdrawals and/or non-evaluable measurements was assumed and, combined with the effect of stratification on analysis, resulted in the requirement to recruit 22 patients per treatment group.
Data analysis
Data are shown as mean±SD. A general linear model (a more general form of ANOVA) was fitted to the data, with treatment group, sex and age strata as the independent factors. When the dependent variable was measured at 24 months, the baseline value was also included as a covariate. In addition, the analyses of all endpoints (e.g. PET and NCS) except blood pressure were adjusted for HbA1c; analyses of nerve conduction data were also adjusted for temperature of the limb. Differences between groups were considered significant at p ≤ 0.05. For the significant tests we also report the estimated adjusted mean difference between the groups; i.e. antioxidant group-adjusted mean change − placebo group-adjusted mean change [20]. We report the analysis using data for participants who completed the trial, i.e. had measurements performed at baseline and 24 months. However, we repeated the analyses of the primary and secondary endpoints with zero differences imputed for participants who did not complete the trial; these analyses gave similar results to the reported analyses. The analyses were performed using SAS 9.2 (SAS Institute, Cary, NC, USA).
Results
Participants
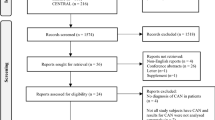
We enrolled 44 participants with type 1 diabetes who met the inclusion criteria and were randomised to either antioxidant treatment or placebo (22 participants per group) (Fig. 1). These participants, 39% of whom were women and 94% of whom were whites, had a mean age of 46 ± 11 years, with 26 ± 12 years duration of diabetes and HbA1c of 58 ± 5 mmol/mol (7.5 ± 1.0%) at baseline. There were no significant differences in any of the demographic or other baseline characteristics of the participants, except for weight, as summarised in Table 1. There were also no differences in HbA1c, systolic or diastolic blood pressure, resting heart rate or total cholesterol between groups at 12 (data not shown) and 24 months of treatment (Table 2). A total of 31 participants completed the 24 month intervention, 18 in the placebo and 13 in the antioxidant group (Fig. 1). The main reasons for dropout are shown in Fig. 1.

Flow diagram of study enrolment. GI, gastrointestinal; SB, small bowel; R, right
Effects of treatment on [11C]HED RI
At baseline there were no differences between groups in global (Table 2) or any regional [11C]HED RI (electronic supplementary material [ESM] Table 1). The antioxidant regimen was associated with a slight, but significant worsening (decreases) of the global [11C]HED RI change compared with placebo after adjusting for age, sex and HbA1c (p = 0.045) (Table 2). No change from baseline was observed with placebo (Table 2).
Analyses of the regional [11C]HED RI also showed that the antioxidant treatment induced slightly greater decreases in the [11C]HED RI in the distal anterior (p = 0.043), proximal lateral (p = 0.043) and proximal anterior (p = 0.03) segments compared with the placebo group (ESM Table 1). In subgroup analyses, no difference was seen in the global or regional [11C]HED RI between participants with a positive change in 24 h urinary F2-isoprostanes (a marker of oxidative stress) and those with a negative change (discussed further below).
Effects of treatment on measures of endothelial function (CFR) and MBF
At baseline and 24 months there were no differences between the antioxidant and placebo groups in the global CFR, a measure of endothelial function and secondary endpoint of this trial (Table 2).
The global and regional resting MBF were similar in participants in both groups at baseline and in the change between baseline and 24 months (ESM Table 1).
No between-group differences were observed in the global stress MBF at baseline (p = 0.48). The global stress MBF was decreased slightly under the antioxidant regimen at 24 months, compared with a slight increase in the placebo group; this difference did not reach statistical significance (p = 0.53). There were no between-group differences in regional stress MBF in the change between baseline and 24 months (ESM Table 1).
Effects of treatment on urinary F2-isoprostanes and CRP
No differences were observed between placebo and antioxidant groups, respectively, in the levels of 24 h urinary F2-isoprostanes at baseline or in the change between baseline and 24 months of treatment (p = 0.87 and p = 0.24, respectively) (Table 2). Similarly, the antioxidant treatment had no effect on CRP levels (Table 2).
Effects of treatment on CARTs, nerve conduction studies and IENFD
No differences were observed at baseline or after treatment for CARTs (Table 2), symptoms and signs of DPN, MNSI findings, NCS, QST or QSART (p = NS for all) (for selected data, see ESM Table 1). IENFD was similar at baseline at the distal leg and at the proximal thigh in the placebo and antioxidant groups, respectively, with no effect of the antioxidant treatment being observed in these measures for the change between baseline and 24 months.
Drug compliance and adverse events
There was no difference between groups in drug compliance (67% active drug group vs 71% placebo, p = 0.68). The majority (89%) of adverse events were of mild-to-moderate severity and most frequently included adverse events related to respiratory, gastrointestinal and musculoskeletal symptoms (Table 3). Together, these three categories represented about 50% of all adverse events. The other 50% was spread over a wide range of categories. Only two serious adverse events occurred, one in each group. Both were of a gastrointestinal nature, and included one death (placebo group).
Discussion
In this cohort of type 1 diabetes patients with mild-to-moderate CAN, an antioxidant regimen designed to affect three different steps in the oxidative stress pathways did not prevent progression of CAN, had no beneficial effects on myocardial perfusion and may have been detrimental (as suggested by the decrease in global [11C]HED RI and MBF). This antioxidant regimen also had no effect on other measures of CAN, such as cardiovascular reflex testing, or on wide-ranging measures of large- and small-fibre neuropathy.
The role of oxidative stress in the pathophysiology of diabetic microvascular complications, including CAN and DPN, has been amply discussed, and the evidence is compelling [3, 5–7, 21]. Moreover, various antioxidants have been shown to prevent or delay the progression of CAN and DPN in animal models [21–24]. For instance, treatment with the antioxidant ALA prevented the formation of reactive oxygen species, caspase-3 activation, nuclear DNA degradation and activation of the receptor for advanced glycation end-products, which have all been shown to promote the development of DPN [25]. The antioxidant allopurinol has been shown to have marked neural and vascular effects in a rat model of diabetes [26] and to attenuate the development of diabetic cardiomyopathy in the streptozotocin-induced mouse model of type 1 diabetes [27]. In another established rat model of type 1 diabetes, nicotinamide was shown to be effective at reversing early DPN [28] and to be neuroprotective in combination with antioxidant melatonin [29]. Furthermore, in the experimental model study that provided a basis for the present clinical trial, our group demonstrated that ALA, allopurinol and nicotinamide had independent effects on oxidative stress and neuronal survival, as well as providing neural protection when used in combination [30].
Our choice of antioxidant regimen was based on the mechanism by which each antioxidant exerts its own effect on attenuating oxidative stress. ALA directly scavenges free radicals, recycles other natural antioxidants, protects peripheral nerves from lipid peroxidation and increases the activity of catalase and superoxide dismutase (SOD) [31], possibly resulting in the normalisation of impaired endoneural blood flow and nerve conduction velocity [31]. Further mechanisms of ALA include: (1) improving the antioxidant defence system through gene expression; (2) inhibiting nuclear factor κB; and (3) activating AMPK in skeletal muscles, with each of these factors having numerous effects [32]. Allopurinol inhibits the xanthine-oxidase pathway, thereby reducing the number of reactive oxygen species formed. Nicotinamide, a water-soluble form of vitamin B3, is a weak first-generation poly (ADP-ribose) polymerase inhibitor and a precursor of NAD+. Besides its antioxidant properties, it has been shown to improve energy status in ischaemic tissue, regulate neuronal calcium fluxes and inhibit apoptosis [28].
Despite the promising results in animal studies, studies of the effects of antioxidant therapy on measures of neuropathy in humans have been disappointing. The conclusions of a 4 month randomised control multicentre trial (DEKAN study) showed that ALA treatment (800 mg/day orally) of type 2 diabetes mellitus patient ‘may slightly improve CAN’ in this patient population [33]. Moreover, the data from seven clinical trials (ALADIN I, II, and III, SYDNEY, SYDNEY 2, ORPIL and NATHAN I) with a total of 1,551 patients treated with ALA (parenteral, oral or combined) for periods ranging from 3 weeks to 4 years are inconclusive [34–37]. Although some studies showed improvements in various symptoms scores, more objective measures were neutral at best. In another study done on 40 adolescents with type 1 diabetes, a controlled-release formulation of ALA did not have any significant effect on markers of oxidative damage or total antioxidant status [38].
A possible explanation for the low rate of success in previous studies is the complexity of mechanisms underlying increased oxidative stress in diabetes, and the fact that using single agents addressing a single pathway have been considered insufficient to effectively correct excess oxidative stress. A recent prospective, non-randomised, open-label study of 50 patients with diabetic neuropathy who were treated with ALA and SOD showed improved nerve conduction velocity and pain perception [39], providing some support for the rationale of our study. However, here we report that our antioxidant approach lacked effectiveness, with no detectable changes in the levels of 24 h urinary F2-isoprostanes, a marker of oxidative stress. This is unlikely to reflect a suboptimal dosing regimen, as therapeutic doses of all agents were used at levels at or above those used in previous clinical reports [34–37, 40]. It is therefore possible that a lack of penetration of the active drug regimen into the target tissues or cellular compartments may have contributed to the lack of efficacy. This raises the possibility that: (1) absorption of these agents was inefficient; and (2) unaccounted for metabolic interactions may have negatively affected the in vivo anti-oxidative properties of the agents when used in combination, possibly nullifying their individual effects. However, with the available data, we were not able to assess causality in this study.
Several other interventional studies using antioxidants have failed, with possible suggested explanations including inadequacy of the doses used, short duration of therapy or poor timing of initiation of the supplement [41]. Another possible reason for the failure of antioxidants to reduce diabetes-related complications is the vast array of mechanisms of glucotoxicity that are independent of oxidative stress. Thus Mooradian and Haas suggest that endoplasmic reticulum stress could explain a recent failure of antioxidant treatment to reduce diabetes-related complications [41].
An additional unexpected observation was the possible detrimental effect of the triple antioxidant on myocardial perfusion. This is in contrast to data obtained in various cohorts of patients with type 1 or type 2 diabetes when using these agents as monotherapy. Allopurinol [42] and nicotinamide [43] used as monotherapies have both been shown to improve endothelial function. However, importantly, allopurinol blocks purine degradation and may result in the accumulation of purine metabolites, including adenosine in the myocardium. This may have contributed to the slightly (although non-significant) higher resting MBF in participants treated with the antioxidant regimen, and to an impaired microvascular responsiveness to adenosine infusion.
Study limitations include the high rate of dropout, the limited power due to the relatively low number of patients, the relatively short duration of the study and the limited assessment of changes in oxidative stress markers. Since this study was designed and initiated, more sensitive techniques such as mass spectrometry have been developed to evaluate multiple oxidative pathway intermediates, which unfortunately, due to sample limitations, were not available for this study. However, this study is to date the first and only one to comprehensively assess the efficacy of a combined antioxidant approach that has been validated in experimental models of neuropathy [30] in participants with diabetes using highly sensitive state of the art technology to characterise CAN, MBF and DPN.
In summary, treatment with a triple antioxidant regimen consisting of ALA, allopurinol and nicotinamide for 24 months failed to prevent changes in oxidative stress and progression of CAN, MBF deficits or DPN in this cohort of patients with type 1 diabetes, suggesting that traditional antioxidant approaches have limited efficacy in human diabetes even when used in combination. These findings are important for the design of future studies using new agents to target pathogenic mechanisms of CAN and DPN development in diabetes.
Abbreviations
- ALA:
-
α-Lipoic acid
- CAN:
-
Cardiovascular autonomic neuropathy
- CARTs:
-
Cardiovascular autonomic reflex tests
- CFR:
-
Coronary flow reserve
- [11C]HED:
-
[11C]Meta-hydroxyephedrine
- CRP:
-
C-reactive protein
- CVD:
-
Cardiovascular disease
- DPN:
-
Diabetic peripheral neuropathy
- IENFD:
-
Intra-epidermal nerve fibre density
- LV:
-
Left ventricle
- MBF:
-
Myocardial blood flow
- MNSI:
-
Michigan Neuropathy Screening Instrument
- NCS:
-
Nerve conduction studies
- PET:
-
Positron emission tomography
- QST:
-
Quantitative sensory testing
- QSART:
-
Quantitative sudomotor axon reflex testing
- RI:
-
Retention index
- SOD:
-
Superoxide dismutase
References
Nathan DM, Cleary PA, Backlund JY et al (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643–2653
Pop-Busui R (2010) Cardiac autonomic neuropathy in diabetes: a clinical perspective. Diabetes Care 33:434–441
Pop-Busui R, Kirkwood I, Schmid H et al (2004) Sympathetic dysfunction in type 1 diabetes: association with impaired myocardial blood flow reserve and diastolic dysfunction. J Am Coll Cardiol 44:2368–2374
Taskiran M, Rasmussen V, Rasmussen B et al (2004) Left ventricular dysfunction in normotensive type 1 diabetic patients: the impact of autonomic neuropathy. Diabet Med 21:524–530
Pennathur S, Heinecke JW (2007) Mechanisms for oxidative stress in diabetic cardiovascular disease. Antioxid Redox Signal 9:955–969
Edwards JL, Vincent AM, Cheng HT, Feldman EL (2008) Diabetic neuropathy: mechanisms to management. Pharmacol Ther 120:1–34
Hur J, Sullivan KA, Schuyler AD et al (2010) Literature-based discovery of diabetes- and ROS-related targets. BMC Med Genomics 3:49
Hoeldtke RD, Bryner KD, VanDyke K (2011) Oxidative stress and autonomic nerve function in early type 1 diabetes. Clin Auton Res 21:19–28
Ziegler D, Gries F (1997) Alpha-lipoic acid in the treatment of diabetic peripheral and cardiac autonomic neuropathy. Diabetes 46:S62–S66
Klein R, Klein BE, Moss SE, Cruickshanks KJ (1998) The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes. Ophthalmology 105:1801–1815
Raffel DM, Wieland DM (2001) Assessment of cardiac sympathetic nerve integrity with positron emission tomography. Nucl Med Biol 28:541–559
Stevens MJ, Raffel DM, Allman KC, Schwaiger M, Wieland DM (1999) Regression and progression of cardiac sympathetic dysinnervation complicating diabetes: an assessment by C-11 hydroxyephedrine and positron emission tomography. Metabolism 48:92–101
Pop-Busui R, Low PA, Waberski BH et al (2009) Effects of prior intensive insulin therapy on cardiac autonomic nervous system function in type 1 diabetes mellitus: the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study (DCCT/EDIC). Circulation 119:2886–2893
Stevens MJ, Dayanikli F, Raffel DM et al (1998) Scintigraphic assessment of regionalized defects in myocardial sympathetic innervation and blood flow regulation in diabetic patients with autonomic neuropathy. J Am Coll Cardiol 31:1575–1584
Feldman EL, Stevens MJ, Thomas PK, Brown MB, Canal N, Greene DA (1994) A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes Care 17:1281–1289
DCCT (1995) Effect of intensive diabetes treatment on nerve conduction in the Diabetes Control and Complications Trial. Ann Neurol 38:869–880
Polydefkis M, Hauer P, Griffin JW, McArthur JC (2001) Skin biopsy as a tool to assess distal small fiber innervation in diabetic neuropathy. Diabetes Technol Ther 3:23–28
Liu W, Morrow JD, Yin H (2009) Quantification of F2-isoprostanes as a reliable index of oxidative stress in vivo using gas chromatography–mass spectrometry (GC-MS) method. Free Radic Biol Med 47:1101–1107
Stevens MJ, Raffel DM, Allman KC et al (1998) Cardiac sympathetic dysinnervation in diabetes: implications for enhanced cardiovascular risk. Circulation 98:961–968
Vickers AJ, Altman DG (2001) Statistics notes: analysing controlled trials with baseline and follow up measurements. BMJ 323:1123–1124
Obrosova IG, Fathallah L, Stevens MJ (2001) Taurine counteracts oxidative stress and nerve growth factor deficit in early experimental diabetic neuropathy. Exp Neurol 172:211–219
Stevens MJ, Obrosova I, Cao X, Van Huysen C, Greene DA (2000) Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes 49:1006–1015
Feldman EL (2003) Oxidative stress and diabetic neuropathy: a new understanding of an old problem. J Clin Invest 111:431–433
Kellogg AP, Wiggin T, Larkin D, Hayes J, Stevens M, Pop-Busui R (2007) Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes 56:2997–3005
Vincent AM, Perrone L, Sullivan KA et al (2007) Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology 148:548–558
Inkster ME, Cotter MA, Cameron NE (2007) Treatment with the xanthine oxidase inhibitor, allopurinol, improves nerve and vascular function in diabetic rats. Eur J Pharmacol 561:63–71
Rajesh M, Mukhopadhyay P, Batkai S et al (2009) Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J Cell Mol Med 13:2330–2341
Stevens MJ, Li F, Drel VR et al (2007) Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J Pharmacol Exp Ther 320:458–464
Negi G, Kumar A, Kaundal RK, Gulati A, Sharma SS (2010) Functional and biochemical evidence indicating beneficial effect of melatonin and nicotinamide alone and in combination in experimental diabetic neuropathy. Neuropharmacology 58:585–592
Vincent AM, Stevens MJ, Backus C, McLean LL, Feldman EL (2005) Cell culture modeling to test therapies against hyperglycemia-mediated oxidative stress and injury. Antioxid Redox Signal 7:1494–1506
Tankova T, Cherninkova S, Koev D (2005) Treatment for diabetic mononeuropathy with alpha-lipoic acid. Int J Clin Pract 59:645–650
Golbidi S, Badran M, Laher I (2011) Diabetes and alpha lipoic acid. Front Pharmacol 2:69
Ziegler D, Schatz H, Conrad F et al (1997) Effects of treatment with the antioxidant α-lipoic acid on cardiac autonomic neuropathy in NIDDM patients. A 4-month randomized controlled multicenter trial (DEKAN Study). Diabetes Care 20:369–373
Ziegler D, Reljanovic M, Mehnert H, Gries FA (1999) Alpha-lipoic acid in the treatment of diabetic polyneuropathy in Germany: current evidence from clinical trials. Exp Clin Endocrinol Diabetes 107:421–430
Ametov AS, Barinov A, Dyck PJ et al (2003) The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid: the SYDNEY trial. Diabetes Care 26:770–776
Ziegler D, Ametov A, Barinov A et al (2006) Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care 29:2365–2370
Ziegler D, Low PA, Litchy WJ et al (2011) Efficacy and safety of antioxidant treatment with alpha-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes Care 34:2054–2060
Huang EA, Gitelman SE (2008) The effect of oral alpha-lipoic acid on oxidative stress in adolescents with type 1 diabetes mellitus. Pediatr Diabetes 9:69–73
Bertolotto F, Massone A (2012) Combination of alpha lipoic acid and superoxide dismutase leads to physiological and symptomatic improvements in diabetic neuropathy. Drugs R D 12:29–34
Ruhnau KJ, Meissner HP, Finn JR et al (1999) Effects of 3-week oral treatment with the antioxidant thioctic acid (alpha-lipoic acid) in symptomatic diabetic polyneuropathy. Diabet Med 16:1040–1043
Mooradian AD, Haas MJ (2011) Glucose-induced endoplasmic reticulum stress is independent of oxidative stress: a mechanistic explanation for the failure of antioxidant therapy in diabetes. Free Radic Biol Med 50:1140–1143
George J, Carr E, Davies J, Belch JJ, Struthers A (2006) High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation 114:2508–2516
Sorrentino SA, Besler C, Rohrer L et al (2010) Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 121:110–122
Funding
The study was funded by the JDRF Centre for the Study of Complications in Diabetes; and by grants P60DK020572 and P30DK092926 to the Michigan Diabetes Research and Training Centre (MDRTC) and the Michigan Center for Diabetes Translational Research (MCDTR) from the National Institute of Diabetes and Digestive and Kidney Diseases.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
RPB performed the study, provided a substantial contribution to the acquisition, analysis and interpretation of data, drafted the article and revised it critically for important intellectual content, and gave final approval of the version to be published. MJS provided a substantial contribution to study conception and design, and to the analysis and interpretation of data, as well as critically revising the manuscript for important intellectual content and giving final approval of the version to be published. DMR provided a substantial contribution to the acquisition, analysis and interpretation of data, revised the manuscript critically for important intellectual content, and gave final approval of the version to be published. EAW and MM provided a substantial contribution to the analysis of data, revised the manuscript critically for important intellectual content and gave final approval of the version to be published. CDP provided a substantial contribution to the acquisition of data, revised the manuscript critically for important intellectual content and gave final approval of the version to be published. MBB analysed all data, provided a substantial contribution to the interpretation of data, revised the manuscript critically for important intellectual content and gave final approval of the version to be published. ELF provided a substantial contribution to study conception and design, revised the manuscript critically for important intellectual content and gave final approval of the version to be published.
Author information
Authors and Affiliations
Corresponding author
Additional information
R. Pop-Busui and M. J. Stevens contributed equally to this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 79 kb)
Rights and permissions
About this article
Cite this article
Pop-Busui, R., Stevens, M.J., Raffel, D.M. et al. Effects of triple antioxidant therapy on measures of cardiovascular autonomic neuropathy and on myocardial blood flow in type 1 diabetes: a randomised controlled trial. Diabetologia 56, 1835–1844 (2013). https://doi.org/10.1007/s00125-013-2942-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2942-9