
Abstract
Aims/hypothesis
The small G-protein ras-related C3 botulinum toxin substrate 1 (RAC1) plays various roles in mammalian cells, such as in the regulation of cytoskeletal organisation, cell adhesion, migration and morphological changes. The present study examines the effects of RAC1 ablation on pancreatic beta cell function.
Methods
Isolated islets from pancreatic beta cell-specific Rac1-knockout (betaRac1 −/−) mice and RAC1 knockdown INS-1 insulinoma cells treated with small interfering RNA were used to investigate insulin secretion and cytoskeletal organisation in pancreatic beta cells.
Results
BetaRac1 −/− mice showed decreased glucose-stimulated insulin secretion, while there were no apparent differences in islet morphology. Isolated islets from the mice had blunted insulin secretion in response to high glucose levels. In RAC1 knockdown INS-1 cells, insulin secretion was also decreased in response to high glucose levels, consistent with the phenotype of betaRac1 −/− mice. Even under high glucose levels, RAC1 knockdown INS-1 cells remained intact with F-actin, which inhibits the recruitment of the insulin granules, resulting in an inhibition of insulin secretion.
Conclusions/interpretation
In RAC1-deficient pancreatic beta cells, F-actin acts as a barrier for insulin granules and reduces glucose-stimulated insulin secretion.
Similar content being viewed by others

Introduction
Type 2 diabetes mellitus is known to be caused by peripheral insulin resistance and pancreatic beta cell failure, and a number of important findings have been made for the molecular mechanism of impaired insulin secretion. The cytoskeleton and cell adhesion of pancreatic beta cells are reported to be involved in insulin secretion, and low-molecular-weight G proteins also play a major role in insulin secretion. About 40 years ago, an electron microscopy study showed that F-actin functions as a barrier for insulin granules [1]. This was the first report to show a relation between the beta cell cytoskeleton and insulin secretion.
Recent studies have found that adjacent beta cells influence each other’s insulin secretion by modulating the F-actin barrier through ephrin receptor (Eph)–ephrin signalling [2]. Such findings suggest that the molecules that regulate the cytoskeleton are important for insulin secretion. In addition, cell culture experiments have also revealed that the amount of insulin secretion is reduced when there is a decrease in the production of cell division cycle 42 (CDC42), which is a low-molecular-weight G protein [3]. Furthermore, RAB27A, member RAS oncogene family (RAB27A) has been shown to play an important role in the docking of insulin granules to the cell membrane [4]. The various roles of small G proteins, including the above examples, in pancreatic beta cells have been well described [5]. In this study, we examined the G-protein ras-related C3 botulinum toxin substrate 1 (RAC1), which plays major roles in various phenomena in the cytoskeleton, in oxidative stress and in secretion. Because of its diverse functions, RAC1 sometimes plays both positive and negative roles in pancreatic beta cells [6]. Previous studies using dominant-negative mutations have shown that inhibiting the functions of RAC1 in cultured beta cells results in decreased insulin secretion [7, 8], suggesting that RAC1 could be an important molecule for insulin secretion; however, no in vivo analyses or studies of the mechanism have been reported to date, and the role of RAC1 at the individual level remains unclear. Here, we conducted a functional analysis of RAC1 in beta cells, using genetically modified mice and cultured cell lines.
Methods
Mice
We generated heterozygous pancreatic beta cell-specific Rac1-knockout (betaRac1 −/−) mice by crossing Rac1 flox/flox mice, which harbour a modified endogenous Rac1 gene in which exon1 is flanked by loxP sites [9], with those that express the Cre recombinase gene under the control of the rat insulin-2 gene [10], as described previously. The mice were backcrossed to mouse strain C57BL/6J for at least eight generations. C57BL/6J mice were obtained from Clea Japan (Tokyo, Japan). Animals were maintained on a 12 h light, 12 h dark cycle and fed normal chow or a high-fat diet containing 30% (wt/wt) fat (14% bovine fat, 14% porcine fat and 2% soybean oil) [11] from the time of weaning (3 weeks old). Blood glucose and plasma insulin concentrations were determined as described previously [12, 13]. All experiments were performed with male mice. This study was performed in accordance with the guidelines of the Animal Ethics Committee of Kobe University Graduate School of Medicine.
OGTT
Mice were deprived of food for 16 h. Blood was collected immediately before and 15, 30, 60 and 120 min after the oral administration of glucose (1.5 mg/g) [14].
Cell culture and transfection of small interfering RNA
At 24 h before transfection, INS-1 cells were re-plated in 12-well plates (60 mm dishes) and transfected with small interfering (si)RNA for Rac1 (SMARTpool; Dharmacon, Lafayette, CO, USA) or scramble controls (Non-Targeting siRNA#2; Dharmacon) with DharmaFECT2 transfection reagent (Dharmacon). After further incubation for 48 h for mRNA, or for 72 h for protein, the cells were harvested for evaluation of insulin secretion and mRNA expression or protein levels.
Assay of insulin secretion from isolated islets
Islets were isolated from 8-week-old mice as described [13, 15]. To assay insulin release, five islets were manually selected, incubated in Krebs–Ringer solution, and stimulated at 37°C for 30 min with various concentrations of glucose, KCl or tolbutamide. The islets were then collected by centrifugation and the supernatant fraction was assayed for insulin secretion using an ELISA kit with a mouse standard (Shibayagi, Gunma, Japan). To measure islet insulin content, islets were solubilised in acid-ethanol solution (74% ethanol, 1.4% HCl [vol./vol.]) overnight at 4°C before insulin ELISA. Insulin secretion from INS-1 cells was measured after a 30 min incubation in Krebs–Ringer–bicarbonate–HEPES buffer (KRBH; 140 mmol/l NaCl, 3.6 mmol/l KCl, 0.5 mmol/l NaH2PO4, 0.5 mmol/l MgSO4, 1.5 mmol/l CaCl2, 2 mmol/l NaHCO3, 10 mmol/l HEPES and 0.1% BSA; pH 7.4) containing the indicated stimulators. The insulin content was determined after extraction with acid ethanol.
Real-time RT-PCR analysis
Total cellular RNA was extracted from INS-1 cells with an RNeasy kit (QIAGEN, Valencia, CA, USA). Real-time RT-PCR analysis of the total RNA extracted from controls and RAC1-knockdown INS-1 cells was performed as described [16]. For real-time quantitative reverse transcription and PCR analysis, cDNA synthesised from total RNA was evaluated in a sequence detector (model 7900; Applied Biosystems, Foster City, CA, USA) with specific primers (electronic supplementary material [ESM] Table 1) and SYBR Green PCR Master Mix (Applied Biosystems). The relative abundance of mRNAs was calculated with cyclophilin mRNA as the invariant control.
Immunoblot analysis
We prepared lysates of isolated islets or INS-1 cells as described previously [12, 13]. The lysates were probed with antibodies to RAC1 (BD Biosciences Pharmingen, San Diego, CA, USA), pan-actin (Cell Signaling, Danvers, MA, USA), and β-actin (Sigma-Aldrich, St Louis, MO, USA).
Immunostaining and morphometric analysis
The pancreas was immersed in Bouin’s solution, embedded in paraffin and sectioned at 4–5 μm thickness. Sections were stained with antibodies to insulin, glucagon (Dako, Glostrup, Denmark), and RAC1 (BD Biosciences). Immune complexes were detected with secondary antibodies conjugated with either cyanine 3 or FITC (Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Quantification of beta cell mass was conducted as described [12, 13].
F-actin staining analysis
F-actin was stained using an F-actin Visualization Biochem Kit (Cytoskeleton, Denver, CO, USA). INS-1 cells cultured on a 12 × 12 mm cover slip were fixed by fixative solution and incubated for 10 min. After washing the cover slip, INS-1 cells were permeabilised by permeabilisation buffer (pH 7.0) and incubated for 5 min. After washing, the F-actin of INS-1 cells was stained with rhodamine phalloidin and incubated for 30 min. For RAC1 staining, an anti-RAC1 antibody (BD Biosciences) was used.
Assay of F-actin/G-actin in vitro
We assayed F-actin/G-actin in INS-1 cells with the G-actin/ F-actin In vivo Assay Kit (Cytoskeleton, Denver, CO, USA). We scraped INS-1 cells in LAS2 buffer (pH 6.9) and lysed them to disrupt the cell membrane. After centrifuging the lysate to a pellet of unbroken cells, we centrifuged the supernatant fraction at 100,000 g to separate F-actin from soluble G-actin. We analysed the supernatant fraction for actin content by immunoblotting with anti-actin antibody.
Perfusion analysis
Perfusion studies of mouse pancreases were performed as described previously [17]. Briefly, overnight-fasted (16 h) male mice (10–12 weeks old) were used. The perfusion protocol began with a 10 min equilibration period using KRBH buffer. The flow rate was 1 ml/min.
Total internal reflection fluorescence microscopy analysis
Mouse pancreatic islets were isolated by the collagenase digestion method and dispersed in Ca2+- and Mg2+-free PBS containing 0.016% trypsin and 0.0066% EDTA at 37°C for 5 min. After washing with ice-cold KRBH, the cells were transferred onto a glass cover slip with high refractive index (refractive index, n 1.8 at 488 nm) (Olympus, Tokyo, Japan) coated with 10 μg/cm2 laminin (Sigma) at 37°C for 3 h. The cells were then infected with adenovirus carrying insulin-Venus at a multiplicity of infection of ten and cultured in DMEM or RPMI-1640 medium supplemented with 10% fetal bovine serum under a humidified condition of 95% air and 5% CO2. After culturing infected primary beta cells for 48 h, the cells on the glass cover slips were pre-incubated on the thermostat-controlled stage at 37°C in KRBH containing 2.8 mmol/l glucose. Then, 30 min after pre-incubation, the cells were incubated with 16.8 mmol/l glucose for 15 min, fixed, immunostained with anti-insulin antibody, and observed by total internal reflection fluorescence microscopy (TIRFM) as described [18, 19].
Statistical analysis
Data are presented as means ± SEM values and compared by ANOVA. A p value of <0.05 was considered significant.
Results
Generation of pancreatic beta cell-specific Rac1-knockout mice
Generalised deficiency of RAC1 in mice has already been reported to be embryonically lethal [20]. Therefore, we generated Rac1-floxed mice with a floxed allele in which exon 1 of Rac1 was sandwiched by loxP [9]. The Rac1-floxed mice were crossbred with Ins-Cre mice that express beta cell-specific Cre, and consequently Rac1 flox/flox, Ins-Cre/+ mice (betaRac1 −/−) deficient in beta cell-specific RAC1 were created, as were Rac1 flox/flox control mice. A western blot confirmed RAC1 production in the pancreatic islets, whole brain, hypothalamus, heart, liver, fats, kidneys and muscles; only the pancreatic islets showed a significant decrease in RAC1 production in betaRac1 −/− mice (Fig. 1a). Immunostaining revealed that RAC1 was produced in both alpha and beta cells in the islets of control mice, while RAC1 was ablated exclusively in pancreatic beta cells of betaRac1 −/− mice (Fig. 1b, c). Ins-Cre mice producing Cre recombinase widely in the brain have recently been reported [21]. However, the clone of the Ins-Cre mouse used for this study did not show any change in expression in the hypothalamus with in situ hybridisation [22], and no difference in blood glucose or insulin level in intraperitoneal glucose tolerance test compared with wild-type mouse [14]. These results indicate that betaRac1 −/− mice are indeed deficient in RAC1, specifically in pancreatic beta cells.

Generation of betaRac1 −/− mice and the effect of RAC1 ablation on glucose metabolism. (a) Immunoblot analysis of RAC1 in islets and in the brain, hypothalamus, heart, liver, fat, kidney and skeletal muscle of control and betaRac1 −/− mice. Immunostaining of pancreas sections of control (b) and betaRac1 −/− (c) mice with antibodies to RAC1, insulin and glucagon. (d–g) Blood glucose and plasma insulin concentrations for control (white bars) and betaRac1 −/− (black bars) mice fed normal chow (d, e) or a high-fat diet (f, g) at the age of 12 weeks. Graphs show the means ± SEM of at least ten control and betaRac1 −/− mice. (h–k) Blood glucose (h, j) and plasma insulin (i, k) concentrations during OGTT are shown for control (n = 6) and betaRac1 −/− (n = 6) mice fed a normal diet (h, i) or a high-fat diet (j, k). * p < 0.05 vs the corresponding value for control mice
BetaRac1 −/− mice showed decreased glucose-stimulated insulin secretion
Mice were reared on a normal diet; body weight, random blood glucose levels and insulin levels were measured in a time-course manner, but no significant differences were found between the knockout and control groups (Fig. 1d, e). Therefore, the mice were reared on a high-fat diet, and their body weight, random blood glucose levels and insulin levels were measured; no differences were found between the two groups (Fig. 1f, g). Next, we performed an OGTT on 8-week-old mice fed a normal diet. In the betaRac1 −/− mice, insulin levels decreased significantly 15 min after the load, accompanied by significant hyperglycaemia 15 min later (Fig. 1h, i). In the same way, an OGTT was performed on 12-week-old mice fed a high-fat diet, and betaRac1 −/− mice showed a decrease in insulin secretion 15 min after the load, with significant hyperglycaemia found 15 min later (Fig. 1j, k).
No change in pancreatic beta cell mass in betaRac1 −/− mice, but insulin secretion was decreased in response to high glucose concentrations
To determine what caused this impaired glucose tolerance, we first examined the impact of RAC1 deficiency on islet morphology and found no significant difference between the two groups in terms of beta cell area or pancreatic islet density (Fig. 2a–d). In addition, no difference in insulin content was found between the two groups (Fig. 2e). Next, to examine the insulin secretory capacity of beta cells, we compared the amount of insulin secreted by isolated pancreatic islets. The amount of insulin secretion in the two groups was not significantly different after low-glucose stimulation, whereas it was significantly decreased in pancreatic islets isolated from the betaRac1 −/− mice after high-glucose stimulation (Fig. 2f). With KCl stimulation, which causes depolarisation of the plasma membrane, no differences were found between the two groups (Fig. 2g). Moreover, the sulfonylurea agent tolbutamide, which induces depolarisation by acting on ATP-dependent K+ channels, was used for stimulation, but no significant differences were found (Fig. 2h). These results revealed that glucose-responsive insulin secretion is impaired when RAC1 is deficient in beta cells.

The effect of RAC1 deficiency on islet characteristics. Immunostaining of pancreatic sections from control (a) and betaRac1 −/− (b) mice at the age of 16 weeks with antibodies to insulin (red) and glucagon (green). Scale bars, 50 μm. (c) Total beta cell mass was calculated from the area of insulin-positive cells in pancreatic sections divided by the total area of the pancreas in 16-week-old control and betaRac1 −/− mice. (d) Islet density was determined from the number of islets in pancreatic sections divided by the total area of the pancreas in 16-week-old control and betaRac1 −/− mice. (e) Insulin content of isolated islets at the age of 16 weeks. (f–h) Insulin release in response to the indicated concentrations of glucose (f), KCl (g) and tolbutamide (h) was measured in islets isolated from control (white bars) or betaRac1 −/− (black bars) mice at the age of 8 weeks. The amount of insulin secreted from the isolated islets is normalised to insulin content. Data were obtained from five independent experiments
Insulin secretion in response to high glucose levels is also decreased in Rac1 knockdown INS-1 cells
For further confirmation of the results, we conducted experiments using a system in which RAC1 was knocked down by loading siRNA into INS-1 cells, a rat-derived beta cell line. Then, RNA was extracted from the cells 48 h after loading the siRNA of Rac1, and proteins were extracted 72 h after loading. Rac1 expression was found to be significantly decreased, both at the protein level and at the mRNA level (Fig. 3a, b). In addition, to confirm that the knockdown generated using siRNA was Rac1-specific, we confirmed the expression of another isoform, ras-related C3 botulinum toxin substrate 2 (RAC2), by using real-time PCR and found that the knockdown has almost no impact on Rac2 expression (Fig. 3b). Rac3 could not be studied because its sequence in rats has not been determined. We measured glucose-responsive insulin secretion by using RAC1 knockdown INS-1 cells and found that high-glucose stimulation resulted in a significant decrease in insulin secretion in the RAC1 knockdown group (Fig. 3c), as is the case in pancreatic islets isolated from mice. However, high potassium stimulation did not result in any significant difference in insulin secretion between the two groups (Fig. 3d). These results suggest that RAC1 contributes to insulin secretion by responding specifically to glucose stimulation.

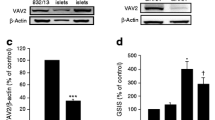
Establishment of RAC1 knockdown INS-1 cells. (a, b) INS-1 cells treated with scramble siRNA (control) and Rac1 siRNA (RAC1 knockdown) were lysed and subjected to immunoblot analysis with antibodies against RAC1 (a) or to real-time PCR analysis of Rac1 and Rac2 mRNA (b). Relative expression values for INS-1 cells treated with scramble siRNA (control) and are means ± SE of three independent experiments. * p < 0.05. (c, d) Insulin secretion in response to the indicated concentrations of glucose or KCl for 30 min was assessed in INS-1 cells and expressed as a proportion of insulin content. Data were obtained from six independent experiments. White bars indicate control, black bars indicate RAC1 knockdown. RAC1 KD, RAC1 knockdown
F-actin remains intact even in the presence of high glucose concentrations because of RAC1 deficiency
To examine the molecular mechanism underlying the decrease in insulin secretion because of a deficiency in RAC1, we performed a pull-down assay using glutathione S-transferase-p21 protein (CDC42/RAC)-activated kinase (GST-PAK), the effector of RAC1, as a substrate and examined the type of insulin secretion stimulation that activates RAC1 in INS-1 cells. Interestingly, RAC1 was activated by stimulation with high concentrations of glucose but not with high concentrations of potassium (Fig. 4a). Quantitative immunoblot analysis revealed that the level of RAC1 activation was significantly higher when stimulated with a high concentration of glucose than when stimulated with a low concentration of glucose or with potassium (Fig. 4a). Thus, RAC1 might be activated only when stimulated by high glucose and might contribute to insulin secretion.

Glucose-stimulated change in F-actin in RAC1-deficient INS-1 cells. (a) INS-1 cells were stimulated with the indicated concentrations of glucose or KCl for 15 min and subjected to the measurement of GTP-RAC1, the active form of RAC1. The bar graph shows quantification of GTP-RAC1 levels. Data are means ± SE of four independent experiments. * p < 0.05, ** p < 0.01. (b–i) Control (b, c, f, g) and RAC1 knockdown (d, e, h, i) INS-1 cells were starved for 30 min in a 2.8 mmol/l glucose solution and then stimulated for 15 min with a 2.8 mmol/l or 25 mmol/l glucose solution. Staining of F-actin in response to the indicated concentrations of glucose was assessed in INS-1 cells treated with scramble siRNA (control) and Rac1 siRNA with rhodamine phalloidin (red) and DAPI (blue) (b–e). Immunostaining of RAC1 in response to the indicated concentrations of glucose was assessed in INS-1 cells treated with scramble siRNA (control) and Rac1 siRNA with antibody to RAC1 (f–i). (j) Immunoblot analysis was performed in 2.8 mmol/l and 25 mmol/l glucose-treated INS-1 cells treated with scramble siRNA (control) and Rac1 siRNA with antibodies to G-actin, F-actin and total actin. The bar graph shows quantification of the G-actin and F-actin levels. Data are means ± SE of five independent experiments. ** p < 0.01. RAC1 KD, RAC1 knockdown
Next, we observed the cytoskeleton of glucose-stimulated INS-1 cells. When stimulated with low concentrations of glucose, the F-actin in the INS-1 cells stained well, but when stimulated with high concentrations of glucose, the amount of stained F-actin decreased significantly. However, when RAC1 was knocked down by siRNA in INS-1 cells, significantly reduced RAC1 production was confirmed by immunostaining (Fig. 4f–i), and F-actin was fully stained, even during high-glucose stimulation, indicating that the F-actin remained significantly more intact in RAC1-deficient cells than in control cells during high-glucose stimulation (Fig. 4b–e). For further confirmation of the results, western blotting was performed to quantify the polymer F-actin and the depolymerised monomer G-actin in the glucose-stimulated INS-1 cells. Under normal conditions, glucose stimulation causes a depolymerisation of F-actin, and consequently the monomer G-actin increases in quantity. However, in the INS-1 cells knocked down for RAC1 by siRNA, the polymer F-actin was present in larger quantities even under glucose stimulation, and no increase in the quantity of G-actin was found (Fig. 4j). Although the proportion of F-actin of the total actin in normal INS-1 cells is reported not to exceed 55% [23, 24], in the present study we found this ratio to be around 70%. This may have been because of the 2.8 mmol/l glucose challenge, which increased the intensity of F-actin polymerisation. Additionally, the toxicity of DMSO [23] and differences in the F-actin assay [24] might have affected the proportion of F-actin. These results suggest that glucose stimulation causes depolymerisation of the F-actin into G-actin in INS-1 cells, but when Rac1 expression is reduced, depolymerisation is inhibited and F-actin remains intact.
RAC1 deficiency inhibits insulin secretion by F-actin
Because the F-actin that has remained intact acts as a barrier in RAC1-deficient cells, it was predicted that the recruitment of insulin granules would be inhibited, causing a decrease in insulin secretion. To examine this hypothesis, the number of insulin granules located at the surface of the beta cell membrane was counted using TIRFM, and the results revealed that, in the beta cells of betaRac1 −/−, the number of insulin granules located at the surface of the beta cell membrane was significantly decreased in comparison to controls (Fig. 5a–c). This might have been because the F-actin that remained intact in the betaRac1 −/− beta cells inhibited the recruitment of insulin granules. In addition, perfusion analysis was performed to examine, at an individual level, the type of influence that RAC1 deficiency in pancreatic beta cells has on the insulin secretion phase (Fig. 5d). The results revealed that the second phase of insulin secretion was significantly decreased in betaRac1 −/− mice (Fig. 5d, e). To confirm that the phenotypes reflected the effect of F-actin, changes in insulin secretion were examined after the cells were loaded with latrunculin B, a reagent that depolymerises F-actin. Latrunculin B also promoted insulin secretion through depolymerisation of F-actin in the beta cells of the control group, but in the betaRac1 −/− cells, a remarkable improvement was found, and there were no differences with the control group (Fig. 5f). The above results indicate that insulin secretion might be reduced during glucose stimulation because the F-actin of pancreatic beta cells functions as a barrier for insulin granules in the betaRac1 −/− cells.

Localisation of insulin granules in RAC1-deficient pancreatic beta cells. (a–c) TIRFM analysis of insulin granule localisation. Comparison of the number of insulin granules docked to the plasma membrane (shown as a highlighted ring) in pancreatic beta cells of control (a) and betaRac1 −/− (b) mice. Primary cultured pancreatic beta cells were pre-incubated with KRBH containing 2.8 mmol/l glucose at 37°C for 30 min, incubated with 16.8 mmol/l glucose for 15 min, fixed, immunostained with anti-insulin antibody, and observed by TIRFM. The surrounding lines represent the outline of a cell attached to the cover glass. The number of docked granules was measured in a cell surface area of 1,000 μm2. Data were obtained from three independent experiments. (d) Glucose-stimulated insulin secretion in vivo was assessed in control (white circles) and betaRac1 −/− mice (black circles) by perfusion analysis. (e) AUC of the first and second phases of insulin secretion in perfusion analysis. (f) Insulin release stimulated with 16.8 mmol/l glucose was assessed with islets from control (white bars) and betaRac1 −/− mice (black bars) pre-treated with or without 10 μmol/l latrunculin B for 30 min. LatB, latrunculin B
Discussion
RAC1 is known to contribute to various disorders, including cancer and neurological disorders [25–27]. In addition, RAC1 is associated with the translocation of GLUT4 via the non-phosphatidylinositol 3-kinase pathway in the muscle of patients with diabetes mellitus [28, 29]. It is also known that many low-molecular-weight G proteins, including RAC1, influence insulin secretion in pancreatic beta cells. Although the importance of the low-molecular-weight G protein in relation to insulin secretion is recognised, RAC1-specific functional analysis has never been performed in pancreatic beta cells. Therefore, we generated betaRac1 −/− mice and analysed them.
It was expected that ablation of RAC1, which mediates cell adhesion and migration, would show morphological abnormalities. Epithelial hyperplasia and a reduced basal cell layer are actually found in epithelial-specific Rac1-knockout mice [30]. However, unexpectedly, no major influence was observed in the pancreatic beta cell mass or in pancreatic islet density in the present study, possibly because other ras-homologous (Rho)-GTPases) compensated to form the pancreatic beta cell mass.
Although some reports have shown that RAC1 contributes to insulin secretion in the pancreatic beta cell line [7, 8], no functional analysis of RAC1 in animals has been performed. This is the first report to evaluate blood glucose levels and serum insulin levels of the betaRac1 −/− mouse. A significant reduction in insulin secretion was found in betaRac1 −/− mice in the OGTT compared with controls. Moreover, significantly reduced insulin secretion was observed in betaRac1 −/− mice when isolated islets were stimulated with glucose. However, no differences in insulin secretion between groups were observed after high KCl or tolbutamide stimulation. Previous reports found no activated RAC1 after high KCl stimulation and showed that when the mutant of inactivated RAC1 was transfected into the cells, glucose-responsive insulin secretion was inhibited without any influence on insulin secretion after high KCl stimulation [7]. In other words, RAC1 in pancreatic beta cells appeared to mediate glucose-specific insulin secretion. Recently, isoprenylcysteine carboxyl methyltransferase (ICMT) was shown to play an important role in glucose-induced RAC1 activation, suggesting that ICMT is a pivotal molecule in glucose-stimulated insulin secretion in pancreatic beta cells [31].
There are many reports that only the first phase of insulin secretion is found after non-nutrient stimulation [32–34]. However, it was not clear which type of molecule and which type of mechanism led to the differences in insulin secretion after glucose stimulation. Our results suggest that the key molecule distinguishing the second phase from the first phase of insulin secretion might have been RAC1. Actually, we demonstrated that only the second phase of insulin secretion was significantly reduced in betaRac1 −/− mice. Previous reports on cultured cell revealed that RAC1 controlled the second phase of insulin secretion [8], which was demonstrated in vivo for the first time in the present study.
In terms of factors regulating the first and second phases of insulin secretion, previous studies have suggested that the readily releasable pool, comprising docked insulin granules constitutes the first phase, and the reserve pool (RP), which is located further away, constitutes the second phase [35]. However, a recently proposed model has the second phase of insulin secretion controlled by an RP formed out of the actin network, irrespective of the distance from the cell membrane or of docking onto the membrane [18]. It is already well known that the actin cytoskeleton plays an important role in insulin secretion [1, 36], and reports have shown that glucose stimulates the recruitment of insulin granules to the cell membrane through actin remodelling [37, 38]. It was recently reported that the Rho–Rho-associated coiled-coil containing protein kinase (ROCK) signalling pathway, in which Rho, another low-molecular-weight G protein, plays an important role, regulates insulin secretion via actin reorganisation in pancreatic beta cells [39]. In our study, with high KCl stimulation and circumstances promoting decreased Rac1 expression, no actin remodelling occurred, suggesting that glucose-stimulated RAC1 activation contributes to F-actin depolymerisation.
In 2007, Konstantinova et al demonstrated that EphA–ephrin-A signalling mediates communication between adjacent pancreatic beta cells, thereby regulating insulin secretion [2]. They showed that EphA–ephrin-A-mediated cell communication is bidirectional, and that EphA forward signalling inhibits insulin secretion while ephrin-A reverse signalling stimulates insulin secretion. They also investigated the downstream targets of EphA–ephrin-A signalling and found that the EphA forward signalling-induced decrease in insulin secretion is accompanied by inhibition of RAC1 activity and an increase in F-actin intensity, while the ephrin-A-reverse-signalling-induced increase in insulin secretion is accompanied by enhanced RAC1 activity with decreased F-actin intensity. However, it remains unclear whether RAC1 activation is linked directly with F-actin intensity, and also with insulin secretion. Here, we demonstrated that activation of RAC1 promotes insulin secretion via depolymerisation of F-actin, suggesting a pivotal role of RAC1 in insulin secretion regulated by communication between adjacent pancreatic beta cells.
Recently, synapses of amphids defective (SAD-A) kinase was identified as a kinase that phosphorylates PAK1, an effector of RAC1 [40]. Although evidence for molecules that act downstream of RAC1 has been slowly accumulating, it is currently not clear how RAC1 regulates the depolymerisation of F-actin. CDC42 was reported to regulate F-actin remodelling in pancreatic beta cell lines [41, 42], and RAC1 acts downstream of CDC42. Both RAC1 and CDC42 regulate the second phase of insulin secretion, and adequate ‘cycling’ of these proteins between the active state (GTP-bound) and the inactive state (GDP-bound) is important in insulin secretion [41, 42]. Considering that inhibition of CDC42 cycling between GTP- and GDP-bound forms impairs insulin secretion and F-actin regulation [41], it is likely that the absence of RAC1 cycling resulted in inhibition of F-actin depolymerisation in the RAC-deficient cells used in the present study. Thus, this study provides evidence for a crucial role of RAC1 cycling in insulin secretion via F-actin depolymerisation in vivo.
GLP-1 secretion in L cells of the small intestine was recently reported to be controlled by the actin barrier, suggesting that low-molecular-weight G proteins such as RAC1 and CDC42 contribute to the molecular mechanism [43]. There are also reports of reduced incretin secretion being found in patients with type 2 diabetes mellitus [44, 45], but the secretion mechanism of incretin remains largely unclear. Interestingly, not only insulin secretion but also incretin secretion might be controlled by RAC1 and CDC42, but further studies are required. Moreover, application of RAC1 to early diagnosis and treatment of diabetes mellitus warrants further investigation.
Abbreviations
- CDC42:
-
Cell division cycle 42
- GST-PAK:
-
Glutathione S-transferase-p21 protein (CDC42/RAC)-activated kinase
- ICMT:
-
Isoprenylcysteine carboxyl methyltransferase
- KRBH:
-
Krebs–Ringer–bicarbonate–HEPES buffer
- MEXT:
-
Ministry of Education, Culture, Sports, Science and Technology (Japan)
- RAC1:
-
Ras-related C3 botulinum toxin substrate 1
- RAC2:
-
Ras-related C3 botulinum toxin substrate 2
- RAB27A:
-
RAB27A, member RAS oncogene family
- Rho-GTPase:
-
Ras-homologous GTPase
- RP:
-
Reserve pool
- Si:
-
Small interfering
- TIRFM:
-
Total internal reflection fluorescence microscopy
References
Orci L, Gabbay KH, Malaisse WJ (1972) Pancreatic beta-cell web: its possible role in insulin secretion. Science 175:1128–1130
Konstantinova I, Nikolova G, Ohara-Imaizumi M et al (2007) EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 129:359–370
Wang Z, Oh E, Thurmond DC (2007) Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem 282:9536–9546
Kasai K, Ohara-Imaizumi M, Takahashi N et al (2005) Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J Clin Invest 115:388–396
Kowluru A (2010) Small G proteins in islet beta-cell function. Endocr Rev 31:52–78
Kowluru A (2011) Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol 81:965–975
Veluthakal R, Kaur H, Goalstone M, Kowluru A (2007) Dominant-negative alpha-subunit of farnesyl- and geranyltransferase inhibits glucose-stimulated, but not KCl-stimulated, insulin secretion in INS 832/13 cells. Diabetes 56:204–210
Li J, Luo R, Kowluru A, Li G (2004) Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am J Physiol Endocrinol Metab 286:E818–E827
Kassai H, Terashima T, Fukaya M et al (2008) Rac1 in cortical projection neurons is selectively required for midline crossing of commissural axonal formation. Eur J Neurosci 28:257–267
Herrera PL (2000) Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127:2317–2322
Inoue H, Ogawa W, Ozaki M et al (2004) Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat Med 10:168–174
Kido Y, Burks DJ, Withers D et al (2000) Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest 105:199–205
Hashimoto N, Kido Y, Uchida T et al (2005) PKClambda regulates glucose-induced insulin secretion through modulation of gene expression in pancreatic beta cells. J Clin Invest 115:138–145
Shigeyama Y, Kobayashi T, Kido Y et al (2008) Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol 28:2971–2979
Kitamura T, Kido Y, Nef S, Merenmies J, Parada LF, Accili D (2001) Preserved pancreatic beta-cell development and function in mice lacking the insulin receptor-related receptor. Mol Cell Biol 21:5624–5630
Matsumoto M, Ogawa W, Akimoto K et al (2003) PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J Clin Invest 112:935–944
Miki T, Minami K, Shinozaki H et al (2005) Distinct effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 on insulin secretion and gut motility. Diabetes 54:1056–1063
Shibasaki T, Takahashi H, Miki T et al (2007) Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci USA 104:19333–19338
Yasuda T, Shibasaki T, Minami K et al (2010) Rim2alpha determines docking and priming states in insulin granule exocytosis. Cell Metab 12:117–129
Sugihara K, Nakatsuji N, Nakamura K et al (1998) Rac1 is required for the formation of three germ layers during gastrulation. Oncogene 17:3427–3433
Wicksteed B, Brissova M, Yan W et al (2010) Conditional gene targeting in mouse pancreatic beta-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59:3090–3098
Hashimoto N, Kido Y, Uchida T et al (2006) Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat Genet 38:589–593
Pigeau GM, Kolic J, Ball BJ et al (2009) Insulin granule recruitment and exocytosis is dependent on p110gamma in insulinoma and human beta-cells. Diabetes 58:2084–2092
Howell SL, Tyhurst M (1980) Regulation of actin polymerization in rat islets of Langerhans. Biochem J 192:381–383
Wang Z, Pedersen E, Basse A et al (2010) Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene 29:3362–3373
Sosa MS, Lopez-Haber C, Yang C et al (2010) Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol Cell 40:877–892
Hayashi-Takagi A, Takaki M, Graziane N et al (2010) Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci 13:327–332
JeBailey L, Wanono O, Niu W, Roessler J, Rudich A, Klip A (2007) Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 56:394–403
Ueda S, Kitazawa S, Ishida K (2010) Crucial role of the small GTPase Rac1 in insulin-stimulated translocation of glucose transporter 4 to the mouse skeletal muscle sarcolemma. FASEB J 24:2254–2261
Benitah SA, Frye M, Glogauer M, Watt FM (2005) Stem cell depletion through epidermal deletion of Rac1. Science 309:933–935
Jayaram B, Syed I, Singh A et al (2011) Isoprenylcysteine carboxyl methyltransferase facilitates glucose-induced Rac1 activation, ROS generation and insulin secretion in INS 832/13 beta-cells. Islets 3:48–57
Curry DL, Bennett LL, Grodsky GM (1968) Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 83:572–584
Rorsman P, Eliasson L, Renstrom E, Gromada J, Barg S, Gopel S (2000) The cell physiology of biphasic insulin secretion. News Physiol Sci 15:72–77
Olofsson CS, Gopel SO, Barg S et al (2002) Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B cells. Pflugers Arch 444:43–51
Rorsman P, Renstrom E (2003) Insulin granule dynamics in pancreatic beta cells. Diabetologia 45:1029–1045
Howell SL, Tyhurst M (1979) Interaction between insulin-granules and F-actin in vitro. Biochem J 178:367–371
Thurmond DC, Gonelle-Gispert C, Furukawa M, Halban PA, Pessin JE (2003) Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethymaleimide-sensitive factor attachment protein receptor protein) complex. Mol Endocrinol 17:732–742
Tomas A, Yermen B, Min L, Pessin JE, Halban PA (2006) Regulation of pancreatic beta-cell insulin secretion by actin cytoskeleton remodelling: role of gelsolin and cooperation with the MAPK signalling pathway. J Cell Sci 119:2156–2167
Hammar E, Tomas A, Bosco D, Halban PA (2009) Role of the Rho-ROCK (Rho-associated kinase) signaling pathway in the regulation of pancreatic beta-cell function. Endocrinology 150:2072–2079
Nie J, Sun C, Faruque O et al (2012) Synapses of Amphids Defective (SAD-A) kinase promotes glucose-stimulated insulin secretion through activation of p21-activated kinase (PAK1) in pancreatic beta-cells. J Biol Chem 287:26435–26444
Nevins AK, Thurmond DC (2003) Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol 285:C698–C710
Wang Z, Thurmond DC (2009) Mechanisms of biphasic insulin-granule exocytosis—roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 122:893–903
Lim GE, Xu M, Sun J, Jin T, Brubaker PL (2009) The rho guanosine 5'-triphosphatase, cell division cycle 42, is required for insulin-induced actin remodeling and glucagon-like peptide-1 secretion in the intestinal endocrine L cell. Endocrinology 150:5249–5261
Toft-Nielsen MB, Damholt MB, Madsbad S (2001) Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 86:3717–3723
Lugari R, Dei Cas A, Ugolotti D et al (2002) Evidence for early impairment of glucagon-like peptide 1-induced insulin secretion in human type 2 (non insulin-dependent) diabetes. Horm Metab Res 34:150–154
Acknowledgements
We thank S. Seino and T. Shibasaki for helpful discussions, and M. Nagano, M. Oya and A. Tanida for technical assistance.
Funding
This work was supported by a grant for CLUSTER from the Ministry of Education, Culture, Sports, Science and Technology (Japan) (MEXT), to M. Kasuga; a grant-in-aid for creative scientific research from MEXT to M. Kasuga (18GS0317), a grant-in-aid for scientific research from MEXT to Y. Kido (22590981), and a Shinryokukai research grant to Y. Kido.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
SA, YS, MS, TH, MM and MK conceived and designed the experiments; SA, YS, KT, HYI, YKa, HE, TM, MKK, NH, WF and HT performed the experiments and acquired the data; SA, SU, TS, HI, AA and YKi analysed and interpreted the data; HYI and YKa contributed to critical revision for important intellectual content. All authors contributed to discussion and reviewed and edited the manuscript. YKi wrote the manuscript. All authors have approved the final version of manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Asahara and Y. Shibutani contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 36 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Asahara, S., Shibutani, Y., Teruyama, K. et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 56, 1088–1097 (2013). https://doi.org/10.1007/s00125-013-2849-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2849-5