
Abstract
Aims/hypothesis
We determined whether: (1) an acute lipid infusion impairs skeletal muscle AMP-activated protein kinase (AMPK)α2 activity, increases inducible nitric oxide synthase (iNOS) and causes peripheral insulin resistance in conscious, unstressed, lean mice; and (2) restoration of AMPKα2 activity during the lipid infusion attenuates the increase in iNOS and reverses the defect in insulin sensitivity in vivo.
Methods
Chow-fed, 18-week-old C57BL/6J male mice were surgically catheterised. After 5 days they received: (1) a 5 h infusion of 5 ml kg−1 h−1 Intralipid + 6 U/h heparin (Lipid treatment) or saline (Control); (2) Lipid treatment or Control, followed by a 2 h hyperinsulinaemic–euglycaemic clamp (insulin clamp; 4 mU kg−1 min−1); and (3) infusion of the AMPK activator, 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) (1 mg kg−1 min−1), or saline during Lipid treatment, followed by a 2 h insulin clamp. In a separate protocol, mice producing a muscle-specific kinase-dead AMPKα2 subunit (α2-KD) underwent an insulin clamp to determine the role of AMPKα2 in insulin-mediated muscle glucose metabolism.
Results
Lipid treatment decreased AMPKα2 activity, increased iNOS abundance/activation and reduced whole-body insulin sensitivity in vivo. AICAR increased AMPKα2 activity twofold; this did not suppress iNOS or improve whole-body or tissue-specific rates of glucose uptake during Lipid treatment. AICAR caused a marked increase in insulin-mediated glycogen synthesis in skeletal muscle. Consistent with this latter result, lean α2-KD mice exhibited impaired insulin-stimulated glycogen synthesis even though muscle glucose uptake was not affected.
Conclusions/interpretation
Acute induction of insulin resistance via lipid infusion in healthy mice impairs AMPKα2, increases iNOS and causes insulin resistance in vivo. However, these changes do not appear to be interrelated. Rather, a functionally active AMPKα2 subunit is required for insulin-stimulated muscle glycogen synthesis.
Similar content being viewed by others


Introduction
Skeletal muscle is the largest organ in the human body and the primary site of insulin-stimulated glucose uptake and metabolism [1]. Any impairment in these processes plays an integral role in the development of metabolic disease states, notably obesity and type 2 diabetes. A major therapeutic option for obesity and type 2 diabetes are the biguanide or thiazolidinedione classes of glucose-lowering drugs, which enhance rates of skeletal muscle glucose uptake and storage [2]. While the exact signalling mechanism(s) responsible for this phenomenon is(are) unclear, a common mechanistic link involves the activation of AMP-activated protein kinase (AMPK), a serine–threonine αβγ heterotrimer thought to act as a regulator of skeletal muscle glucose fluxes [3]. Indeed, the metal binding properties of biguanides appear to regulate the activation of AMPK [4].
Pharmacological activation of AMPKα2 is associated with improved insulin sensitivity [5–7]. The mechanism(s) by which AMPKα2 mediates this process is(are) unclear, although one possible mechanism relates to suppression of inducible nitric oxide synthase (iNOS). iNOS, an isozyme of the nitric oxide synthase family, plays a role in a number of biological processes, including pathological conditions such as insulin resistance. In skeletal muscle, iNOS abundance is enhanced in obesity and type 2 diabetes [8, 9]. Deletion of iNOS improves insulin sensitivity [8, 10] and intracellular insulin signalling [11] in animal models of insulin resistance. In cell cultures, reduced AMPKα activation is associated with enhanced iNOS production [12], whereas pharmacological activation of AMPKα reduces iNOS levels [12, 13]. Obesity and insulin resistance are associated with impaired skeletal muscle AMPKα2 activation [6, 14, 15], although not all studies have observed this [16, 17]. Nevertheless, it is possible that the impaired skeletal muscle AMPKα2 activation seen in obesity and type 2 diabetes contributes to the elevated iNOS levels seen in these metabolic disease states. Due to the many complexities involved in obesity and type 2 diabetes, the association between AMPKα2 and iNOS is difficult to pinpoint.
The aim of this study was to elucidate the role of skeletal muscle AMPKα2 in insulin resistance, without the associated complications of chronic obesity. Specifically, we tested the hypothesis that: (1) an acute lipid emulsion infusion that results in insulin resistance would impair skeletal muscle AMPKα2 activity in vivo in lean healthy mice; (2) impaired AMPKα2 activity would lead to enhanced levels of iNOS in skeletal muscle; and (3) the restoration of AMPKα2 activity during the lipid infusion (via infusion of the AMPK activator AICAR) would reverse the defect in insulin sensitivity in vivo by suppressing iNOS.
Methods
Animal maintenance
The principles of laboratory animal care were followed. Procedures were approved by the Vanderbilt University Animal Care and Use Committee. Male, 18-week-old C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME, USA) and 16-week-old C57BL/6 mice expressing a muscle-specific kinase-dead Ampkα2 (also known as Prkaa2) subunit (α2-KD) [18] were used. All mice were fed a standard chow diet (5001 Laboratory Rodent Diet; Purina, Richmond, IN, USA) with free access to water.
Surgical procedures
At 5 days prior to the experiment, mice underwent surgery to catheterise the left common carotid artery and right jugular vein as described previously [19], with the modification that mice were anaesthetised with isofluorane. Mice were housed individually after surgery and were within 10% of the pre-surgery weight on the day of study.
Intralipid infusion and hyperinsulinaemic–euglycaemic clamp protocols
Between 07:00 and 08:00 hours mice were placed into new cages containing bedding, but no food and water, and underwent one of four protocols (Fig. 1) as follows.
-
Protocol 1: lipid emulsion (Lipid treatment; 5 ml kg−1 h−1 Intralipid + 6 U/h heparin) or an equivalent volume of saline (Control) was infused for 5 h (t = −300 to 0 min). Arterial blood samples were taken hourly. At t = 0 min, mice were killed with pentobarbital sodium, and skeletal muscle groups were excised, snap-frozen in liquid nitrogen and stored at −80°C.
-
Protocol 2: mice received Lipid treatment or Control as above, followed by a 2 h hyperinsulinaemic–euglycaemic clamp (insulin clamp; 4 mU kg−1 min−1 [19]) beginning at t = 0 min. Lipid or Control treatments were continued throughout the insulin clamp. To prevent a fall of >5% in packed cell volume during the insulin clamp, mice received saline-washed erythrocytes from donors throughout the experimental period (1 μl/min from t = −240 to 0 min; 4 μl/min from t = 0 to 120 min). Following the final blood sample, mice were anaesthetised and killed, and tissues were collected.
-
Protocol 3: mice received Lipid treatment for 5 h. At t = −240 min mice also received a continuous infusion of AICAR (1 mg kg−1 min−1) (Toronto Research Chemicals, Toronto, ON, Canada) or an equal volume of saline as a control. At t = 0 min mice underwent a 2 h insulin clamp as in Protocol 2, with Lipid treatment and AICAR or saline infusion continuing throughout the insulin clamp. After the final blood sample, 50 μl yellow Dye-Trak microspheres (15 μm; Triton Technology, San Diego, CA, USA) were injected into the carotid artery, followed by a small flush of saline, to assess per cent of cardiac output to gastrocnemius muscle (%QG). Mice were anaesthetised and killed, and tissues were collected. In a subset of mice, rates of whole-body and tissue-specific glucose fluxes were also determined using [3-3H]glucose and 2-deoxy[14C]glucose (2-[14C]DG), respectively [19, 20]. To determine endogenous (Endo) rates of glucose appearance (R a) (EndoR a) and glucose disappearance (R d), a 74 kBq bolus of [3-3H]glucose was administered at t = −180 min, followed by a continuous 5.9 kBq/min infusion of [3-3H]glucose. At t = 78 min, a 481 kBq bolus of 2-[14C]DG was injected into the jugular vein to determine tissue-specific indices of muscle glucose uptake (R g).
-
Protocol 4: mice received Lipid treatment for 5 h. At t = −90 min the insulin clamp commenced and at t = 0 min AICAR (1 mg kg−1 min−1) was infused for 2 h. Lipid treatment was infused throughout this period. EndoR a and R d were determined. Saline-washed erythrocytes were given as for Protocol 2.

Schematic outlines of the four different infusion protocols used in C57BL/6J wild-type mice, i.e. (a) Protocol 1, (b) Protocol 2, (c) Protocol 3 and (d) Protocol 4. Arrow (c), bolus administration of 2-[14C]DG at 78 min
In a separate study, α2-KD mice that had fasted for 5 h underwent a standard 2 h insulin clamp [19] to determine insulin-stimulated glycogen synthesis.
Plasma analyses
Arterial glucose was determined from ∼5 μl arterial blood using a monitor (Accu-Chek Advantage; Roche Diagnostics, Indianapolis, IN, USA). Immunoreactive plasma insulin was assayed with a double antibody method [21]. Plasma NEFA were measured spectrophotometrically (NEFA C kit; Wako Chemicals, Richmond, VA, USA). Plasma lactate was measured enzymatically [20]. Plasma for [3-3H]glucose and 2-[14C]DG measurements was deproteinised with Ba(OH)2 and ZnSO4, and an aliquot was counted either directly or after being dried to remove 3H2O [19].
Tissue preparation
Whole-cell lysates from the gastrocnemius muscle were prepared as described [20]. Protein content was determined using the Bradford method. Following extraction with perchloric acid, adenine nucleotides were measured by HPLC [22] and glucose-6-phosphate via enzymatic methods [23]. Tissue samples for 2-[14C]DG and 2-[14C]DG-phosphate were determined as described [20]. Glycogen was determined using the method of Chan and Exton [24].
Assays
AMPKα2 and -α1 activity was measured following immunoprecipitation as described [20]. AMPK activity in the immune complexes was measured in the presence of 200 μmol/l AMP and calculated as incorporation of phosphate into the SAMS peptide (100 μmol/l; GenWay Biotech, San Diego, CA, USA), expressed as pmol min−1 mg−1 of protein subjected to immunoprecipitation. iNOS activity was measured as described previously for total NOS activity [20], except that EGTA (1 mmol/l) was added to the assay buffer to inhibit Ca2+-dependent NOS activity (i.e. neuronal and endothelial NOS isozymes) and the assay was performed for 25 min at 37°C. iNOS activity was calculated as conversion of l-[3H]arginine to l-[3H]citrulline in pmol min−1 mg−1 of protein. Glycogen synthase (GS) and glycogen phosphorylase activity was assessed as described [25].
Immunoblotting
AMPKα2 protein levels and acetyl-CoA carboxylase (ACC)β abundance/Ser221 phosphorylation were determined from 100 μg whole-cell lysate. Proteins were separated using gels (NuPAGE 4-12% (vol./vol.) Bis-Tris Gels; Invitrogen, Carlsbad, CA, USA). Blots were probed with an AMPKα2 goat polyclonal (Santa Cruz, Santa Cruz, CA, USA) or ACCβ Ser221 rabbit polyclonal antibody (Cell Signaling, MA, USA), respectively. ACCβ protein abundance was detected using IRDye 800-labelled streptavidin (Rockland Immunochemicals, Gilbertsville, PA, USA). Protein levels were normalised to GAPDH (glyceraldehyde 3-phosphate dehydrogenase; Abcam, Cambridge, MA, USA). Antibody binding for AMPKα2 and ACCβ Ser221 was detected with an IRDye 800-conjugated anti-goat or anti-rabbit IgG. An IRDye 700-conjugated anti-mouse IgG secondary antibody (Rockland Immunochemicals) was used for GAPDH. iNOS abundance was determined in Protocol 1 by immunoprecipitation of iNOS on 500 μg protein using Recomb protein A beads and 2 μg of a mouse iNOS monoclonal antibody (BD Biosciences, San Jose, CA, USA). In Protocol 3, iNOS abundance was determined from 100 μg whole-cell lysate.
Calculations
EndoR a and R d were determined using Steele's non-steady-state equations [26]. EndoR a was determined by subtracting the exogenous glucose infusion rate (GIR) from total R a. Whole-body glycolytic rates were estimated from the increment per unit time of 3H2O multiplied by estimated body water (assumed to be 60% of body weight) and divided by [3-3H]glucose-specific activity. The appearance of 3H2O was determined by linear regression of samples from t = 80 to 120 min. The whole-body glycogen synthesis rate was calculated as the R d minus the whole-body glycolytic rate. To determine insulin-stimulated muscle glycogen synthesis, the specific activity of gastrocnemius [3-3H]glucose was multiplied by gastrocnemius glycogen content and divided by the specific activity of plasma [3-3H]glucose. Indices of tissue-specific clearance of 2-[14C]DG (K g) and R g were calculated as described [27]. Per cent of cardiac output to gastrocnemius muscle was calculated as described [20, 28].
Statistical analyses
Data are mean ± SEM. Statistical analysis was performed using a Student's t test, one-way ANOVA or two-way repeated measures ANOVA where appropriate, with the statistical software package SigmaStat (Systat Software, San Jose, CA, USA). If the ANOVA was significant (p < 0.05), specific differences were located using Fisher's least significant difference (LSD) test.
Results
Acute lipid-induced insulin resistance impairs skeletal muscle AMPKα2 activation and enhances iNOS levels
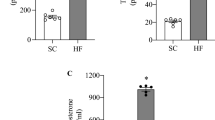
After 1 h of lipid treatment (Protocol 1; Fig. 1a), plasma NEFA levels were significantly elevated compared with Control (2.7 ± 0.5 vs 1.2 ± 0.2 mmol/l, respectively; p < 0.05) and remained elevated over the subsequent 4 h (Fig. 2a). Lipid treatment did not alter arterial glucose (6.0 ± 0.4 vs 5.7 ± 0.3 mmol/l for Control) or plasma insulin levels (0.46 ± 0.03 vs 0.36 ± 0.10 nmol/l, respectively). Lipid treatment reduced gastrocnemius AMPKα2 activity by 25 ± 7% compared with Control (p < 0.05) (Fig. 2b). This was not due to alterations in AMPKα2 protein levels (82 ± 4 vs 83 ± 2 arbitrary units for Control) or adenine nucleotide levels (Table 1). Gastrocnemius AMPKα1 activity (Fig. 2b) and ACCβ Ser221 phosphorylation (Fig. 3f) were not significantly altered. Lipid treatment increased gastrocnemius iNOS abundance (Fig. 2c) and activity (Fig. 2d) ∼1.5-fold.

Effect of a 5 h infusion of 5 ml kg−1 h−1 Intralipid + 6 U/h heparin (Lipid treatment) or of an equivalent volume of saline (Control) on (a) plasma NEFA levels, (b) gastrocnemius AMPKα2 and -α1 activity, (c) iNOS abundance (as fold change vs control) and (d) iNOS activity. Data are mean ± SEM for n = 5 per group; *p < 0.05 vs corresponding control value

(a) Plasma NEFA levels, (b) arterial glucose levels and (c) GIR prior to (−300 to 0 min) and during (0 to 120 min) a 2 h hyperinsulinaemic–euglycaemic clamp as described in the Methods, in mice that received 5 ml kg−1 h−1 Intralipid + 6 U/h heparin from −300 to 120 min. White circles, Lipid treatment; black circles, Control. (d) Gastrocnemius AMPKα2 activity, (e) AMPKα1 activity and (f) ACCβ Ser221 phosphorylation. Data are mean ± SEM for n = 5–9 per group; *p < 0.05 vs corresponding Control value; † p < 0.05 vs corresponding non-insulin value
Plasma NEFA levels remained elevated throughout the subsequent 2 h insulin clamp (Protocol 2; Fig. 1b) in Lipid treatment mice (Fig. 3a), which were also insulin-resistant, as evidenced by a 51 ± 6% reduction in the GIR required to maintain euglycaemia during the insulin clamp (Fig. 3b, c). This occurred despite significantly higher plasma insulin levels (2.1 ± 0.3 vs 1.0 ± 0.1 nmol/l for Control; p < 0.001). In Control mice, insulin markedly suppressed AMPKα2 and -α1 activity (Fig. 3d, e), and ACCβ Ser221 phosphorylation (Fig. 3f). Insulin suppressed AMPKα2 activity in Lipid treatment mice (Fig. 3d), although the relative suppression was attenuated compared with Control mice (45 ± 3 vs 63 ± 3%, respectively; p = 0.001). Insulin did not suppress AMPKα1 activity in Lipid treatment mice (Fig. 3e), while ACCβ Ser221 phosphorylation was suppressed to similar levels to those in Control mice (Fig. 3f).
AICAR increases skeletal muscle AMPKα2 activity
Given that Lipid treatment-induced impairment of skeletal muscle AMPK signalling was associated with insulin resistance and enhanced iNOS activation, we determined whether activation of AMPKα2 (via the AMPK activator AICAR) during lipid infusion (Protocol 3; Fig. 1c) could suppress iNOS activation, thus improving insulin sensitivity. AICAR was infused at 1 mg kg−1 min−1 for the last 4 h of the Lipid treatment (t = −240 to 0 min) and continued throughout the 2 h insulin clamp (t = 0–120 min). Previous studies in rodents and humans assessing AICAR-stimulated muscle glucose metabolism in vivo have used doses ranging from 0.125 to 10 mg kg−1 min−1 [29–34]. A dose of 1 mg kg−1 min−1 required an infusion of exogenous glucose to maintain arterial glucose at levels similar to those in saline-infused mice. Preliminary experiments confirmed that the dose of exogenous glucose required at 1 mg kg−1 min−1 AICAR (glucose 10 ± 3 mg kg−1 min−1, n = 7) did not differ from that required at 10 mg kg−1 min−1 AICAR (glucose 13 ± 2 mg kg−1 min−1, n = 9).
Gastrocnemius ATP, ADP and AMP levels were not altered in response to AICAR by the end of the insulin clamp (Fig. 4a). However, 5-amino-4-imidazolecarboxamide ribotide (ZMP) levels increased to ∼75% of AMP levels (Fig. 4b). In line with increased ZMP, gastrocnemius AMPKα2 activity (Fig. 4c) and ACCβ Ser221 phosphorylation (Fig. 4d) were ∼2.5-fold greater at the end of the insulin clamp in AICAR-infused mice. AMPKα1 activity remained unchanged (data not shown). The increase in AMPKα2 activity did not impact on iNOS levels (Fig. 4e) or iNOS activity (Fig. 4f).

AICAR (1 mg kg−1 min−1) (white bars) or an equivalent volume of saline (black bars) was infused during the last 4 h of a 5 h lipid infusion (5 ml kg−1 h−1 Intralipid + 6 U/h heparin) and a subsequent 2 h insulin clamp. (a, b) Gastrocnemius adenine nucleotide levels, (c) AMPKα2 activity, (d) ACCβ Ser221 phosphorylation (relative to ACCβ), (e) iNOS abundance (relative to GAPDH) and (f) iNOS activity were determined. Dashed line (c, d, f), values in mice following 5 h of Lipid treatment or Control (i.e. in the absence of insulin). Data are mean ± SEM for n = 11–12 (a, b) and n = 5–7 (c–f); *p < 0.05 vs corresponding saline value
AICAR infusion does not improve whole-body insulin sensitivity during acute hyperlipidaemia
Infusion of AICAR during Lipid treatment (i.e. t = −240 to 0 min) did not alter plasma insulin levels (Fig. 5a). However, AICAR did require an exogenous glucose infusion, with 10 ± 3 mg kg−1 min−1 needed at t = 0 min to maintain glucose at similar levels to those in saline-infused mice (Fig. 5b, c). The GIR requirement in response to AICAR infusion was due to suppressed EndoR a (Fig. 5d), as opposed to altered R d, which was unchanged in AICAR- vs saline-infused mice (Fig. 5e). AICAR infusion also significantly increased plasma lactate levels (Fig. 5f).

Plasma insulin levels (a), blood glucose levels (b) and GIR, with insulin-induced change in GIR (∆GIR) (c) during a 2 h insulin clamp, which was preceded by a 5 h lipid infusion (5 ml kg−1 h−1 Intralipid + 6 U/h heparin) in conjunction with AICAR (1 mg kg−1 min−1) (white circles) or an equivalent volume of saline (black circles), which was infused during the last 4 h of the lipid infusion. The lipid and AICAR or saline infusions were continued throughout the clamp. (d) The effect of the above treatment on EndoR a, (e) R d and (f) plasma lactate levels. Data are mean ± SEM for n = 11–12; *p < 0.05 for main effect, AICAR; † p < 0.05 for main effect, insulin
AICAR infusion prior to and during the 2 h insulin clamp did not alter insulin-stimulated GIR (Fig. 5c). Insulin as such suppressed EndoR a, while AICAR plus insulin exerted an additive effect on EndoR a suppression, resulting in reduced absolute rates of EndoR a at the end of the insulin clamp compared with saline-infused mice (Fig. 5d). However, the insulin-stimulated reduction of EndoR a (i.e. from t = 0–120 min) was similar between AICAR- and saline-infused mice (decreases of 15 ± 3 vs 13 ± 5 mg kg−1 min−1, respectively). Insulin increased R d (Fig. 5e), however no additive effect of AICAR was observed. Plasma lactate levels at the end of the insulin clamp remained elevated in AICAR-infused mice (Fig. 5f). AICAR did not alter the per cent cardiac output to gastrocnemius muscle at the end of the insulin clamp (data not shown).
The results described above suggest that a pharmacological increase in skeletal muscle AMPKα2 activity (via AICAR) is not sufficient to induce alterations in whole-body glucose disposal (i.e. R d). To rule out the possibility that the order of AICAR and insulin infusions was a factor in determining results, a subset of mice underwent an insulin clamp that commenced during the last 90 min of Lipid treatment (i.e. t = −90 to 0 min), followed by a 2 h AICAR infusion (i.e. t = 0–120 min; Protocol 4; Fig. 1d). Insulin infusion from t = −90 to 0 min increased GIR to 40 ± 2 mg kg−1 min−1 and increased R d from 20 ± 1 to 35 ± 2 mg kg−1 min−1 (p < 0.01). However, the addition of AICAR from t = 0–120 min did not further alter GIR (42 ± 3 mg kg−1 min−1) or R d (36 ± 3 mg kg−1 min−1) compared with the preceding 90 min insulin infusion.
AICAR infusion in vivo increases rates of insulin-stimulated glycogen synthesis
Despite no change being observed in R d, whole-body glycolytic rates, interestingly, were suppressed during the 4 h AICAR infusion (i.e. t = −240 to 0 min), while glycogen synthesis rates were elevated (Fig. 6a, b). Insulin suppressed glycolytic rates and increased synthesis rates regardless of AICAR or saline infusion. However, glycolysis remained attenuated and synthesis enhanced in mice receiving AICAR and insulin. Likewise, gastrocnemius glycogen levels were significantly elevated in AICAR- plus insulin-infused mice (Fig. 6c), as was the rate of [3-3H]glucose incorporated into muscle glycogen (i.e. enhanced glycogen synthesis) (Fig. 6d). However, gastrocnemius R g did not differ between AICAR plus insulin and saline plus insulin mice (Fig. 6e), findings also observed in other muscle groups (data not shown). Glucose-6-phosphate levels were elevated ∼30% in mice infused with AICAR and insulin (p = 0.1) (Fig. 6f), while GS (Fig. 6g) and glycogen phosphorylase (Fig. 6h) activities were unchanged.

The effect of a 4 h AICAR infusion (1 mg kg−1 min−1) or an equivalent volume of saline, followed by a 2 h insulin clamp on (a) whole-body rates of glycolysis and (b) glycogen synthesis, expressed as a fraction of whole-body R d. Lipid treatment at 5 ml kg−1 h−1 Intralipid + 6 U/h heparin was administered throughout. (c) Also shown are gastrocnemius glycogen content, (d) [3-3H]glucose incorporation into glycogen, (e) the glucose metabolic index (R g), (f) glucose-6-phosphate (G-6-P) levels, (g) GS activity and (h) glycogen phosphorylase (GPh) activity under the above conditions. Data are mean ± SEM for n = 6 per group; *p < 0.05 for main effect, AICAR; † p < 0.05 for main effect, insulin; ‡ p < 0.05 vs corresponding saline value
A kinase-dead AMPKα2 subunit in skeletal muscle impairs insulin-mediated glycogen synthesis in vivo
The above data indicated a role for AMPKα2 in determining the cellular fate of glucose. To determine whether activation of AMPKα2, as opposed to an AMPKα2-independent effect of AICAR, accentuated insulin-stimulated glycogen synthesis, lean, otherwise healthy α2-KD mice underwent an insulin clamp. Such α2-KD mice have essentially no skeletal muscle AMPKα2 activity at rest or in response to physiological stimuli such as exercise [20]. Interestingly, iNOS activity was significantly elevated in α2-KD mice following a 5 h fast (2.2 ± 0.4 vs 0.5 ± 0.3 pmol min−1 mg−1 for wild-type; p < 0.02).
No differences between α2-KD and wild-type mice were observed with respect to blood glucose, GIR, EndoR a, and R d during the insulin clamp (Fig. 7a–d). However, in α2-KD mice, skeletal muscle glycogen was significantly lower (Fig. 7e), the rate of [3-3H]glucose incorporation into skeletal muscle glycogen was reduced by 47 ± 10% (Fig. 7f) and there was a strong tendency for reduced rates of whole-body glycogen synthesis (0.35 ± 0.07 vs 0.51 ± 0.05 for wild-type; p = 0.08). Thus, a functionally inactive AMPKα2 subunit in skeletal muscle is associated with impaired insulin-stimulated glycogen synthesis.

Blood glucose (a), GIR (b), EndoR a (c) and R d (d) before (Basal) and in response to an insulin clamp (0 to 120 min) in wild-type (WT) mice (white circles/bars) and mice producing α2-KD in skeletal muscle (black circles/bars). (e) Glycogen levels following the insulin clamp and (f) glycogen synthesis during the insulin clamp. Data are mean ± SEM for n = 5–6; *p < 0.05 vs corresponding WT value; † p < 0.05 vs Basal; ‡ p = 0.05
Discussion
Obesity and insulin resistance are associated with impaired AMPKα2 activation [14] and enhanced iNOS abundance/activation [9] in skeletal muscle. Direct evidence for an AMPKα2–iNOS axis is unclear and the ability to test a casual relationship is confounded by the many complications associated with these chronic metabolic disease states. Here, we show that in healthy mice, the induction of acute hyperlipidaemia reduces skeletal muscle AMPKα2 activity and increases iNOS levels/activation in vivo. However, a reduction of AMPKα2 activity is not required for the lipid-induced increase of iNOS. Nor is an active AMPKα2 subunit required for improved insulin-stimulated muscle glucose uptake or whole-body insulin action. Indeed, rescue of the lipid-induced impairment of AMPKα2 activity via AICAR infusion did not improve whole-body insulin-stimulated glucose uptake. Rather, in lipid-infused mice, pharmacological activation of AMPKα2 in skeletal muscle was associated with an increase in the percentage of glucose taken up and directed into glycogen under insulin-stimulated conditions. This finding was supported by the demonstration of impaired insulin-stimulated glycogen synthesis, but not glucose uptake, in lean mice expressing a dominant-negative mutation of the Ampkα2 subunit in skeletal muscle.
A major question arising from our collective studies is how AICAR can alter muscle glycogen synthesis independently of alterations in key determinants of glucose uptake and glycogen synthesis and degradation. Our finding is not unique. In high fat-fed rats, the injection of AICAR (250 mg/kg) 24 h prior to an insulin clamp did not alter insulin-stimulated R d or R g in red quadriceps muscle, yet red quadriceps glycogen synthesis increased [5]. In that study, glucose-6-phosphate, GS and glycogen phosphorylase were not assessed. While we do not have conclusive evidence, we believe that the effect of AICAR on the modulation of muscle glycogen synthesis is mediated via AMPKα2. It has been shown that the addition of AICAR (2 mmol/l) to rat muscle made insulin-resistant via palmitate incubation improved insulin-stimulated glycogen synthesis (6 nmol/l insulin) in vitro, an effect which was ablated in the presence of an AMPK inhibitor [35]. Nevertheless, in that study [35] AICAR did induce a small, albeit significant increase in insulin-stimulated glucose transport at 6 nmol/l insulin, and a ∼65% increase in glucose-6-phosphate levels. Glucose-6-phosphate levels were elevated ∼30% in our study, and while this was not statistically significant (p = 0.1), one cannot discount the possibility that this increase is enough to elicit a change in intramuscular glucose metabolism in vivo. Interestingly, AICAR also increases glycogen synthesis in insulin-resistant muscle in vitro in response to 0.6 nmol/l insulin [35]. Unfortunately, glucose-6-phosphate levels and glucose transport rates were not assessed at this tenfold lower insulin dose, although one would expect these variables to be attenuated.
It cannot be ruled out that our findings are in part due to off-target effects of AICAR. Other, more specific AMPK activators are now available (e.g. A-769662). However, it has been shown that this compound is selective for AMPKβ1 heterotrimeric complexes, which represent a small fraction of AMPK heterotrimers in skeletal muscle [36]. As with AICAR, off-target effects of A-769662 have also been observed [36]. Alternatively, while the majority of evidence suggests that AICAR–AMPKα2 is not involved in the regulation of glycogen phosphorylase [37, 38], it is possible that AICAR–AMPKα2 altered the targeting of GS. However, in insulin-resistant muscle, activation of AMPK via AICAR does not alter GS activity or key GS phosphorylation sites [35]. Similarly, the total activity of protein phosphatase-1, a major glycogen targeting subunit [39], appears to be unaltered in response to an Intralipid infusion [40].
A key difference between our current study and almost all previous in vivo studies is the dose of AICAR infused and also the plasma insulin concentration. This is not a trivial point. Thus here we observed increased AMPKα2 activity at 1 mg kg−1 min−1 AICAR, a much lower dose than the 7.5 to 10 mg kg−1 min−1 used by others [29, 30, 33], who also observed elevated R d and muscle glucose uptake. Given that AICAR increases ZMP (an AMP analogue) and thus ZMP:ATP, it is possible that the elevated rates of muscle glucose uptake seen with higher AICAR doses are at least partially AMPKα2-independent. An example of this relates to gene transcription. Stoppani et al [34] saw increased AMPKα2 activation and enhanced transcription of uncoupling protein 3 and hexokinase II in response to 1,000 mg/kg AICAR, an effect that was accompanied by hypoglycaemia and elevated blood lactate levels. Reducing the total AICAR dose to 30 mg/kg still increased skeletal muscle AMPKα2 activation and transcription of uncoupling protein 3 and hexokinase II, but did not alter blood glucose or lactate levels [34]. Similarly, it has been shown that AICAR-, but not insulin-stimulated glycogen synthesis is impaired in α2-KD mice in vitro [38]. This differs from our current finding, whereby α2-KD mice exhibited a reduction of insulin-stimulated glycogen synthesis. However, Hunter and colleagues incubated muscles with 60 nmol/l of insulin [38], well above the higher end of the physiological range used in the present study (average of ∼1.5 nmol/l across all studies).
Our findings are consistent with studies showing that: (1) a constitutively active AMPKα2 subunit (via a R225Q mutation in Ampkγ3 [also known as Prkag3]) is associated with elevated muscle glycogen levels following feeding, fasting or the post-exercise period [41, 42]; and (2) 10 weeks of metformin treatment increases basal AMPKα2 activity and muscle glycogen levels in humans [7]. It should also be noted that an acute Intralipid infusion, shown here to reduce AMPKα2 activity, also reduces skeletal muscle glycogen synthesis [43]. Our finding that skeletal muscle glycogen synthesis is impaired in α2-KD mice demonstrates that AMPKα2 plays a role in this process. However, our findings differ from those of Viollet et al [44], who found impaired muscle glycogen synthesis in Ampkα2 −/− mice (global deletion of Ampkα2), but not in α2-KD mice (muscle-specific mutation) during an in vivo insulin clamp. The reason for this difference is unclear, especially when considering that basal GS activity in vitro is markedly increased in skeletal muscle of Ampkα2 −/− [45], but not in α2-KD mice [18].
Skeletal muscle iNOS activation is enhanced under conditions of chronic obesity and insulin resistance [8, 9], while activation of AMPKα2 via AICAR has been shown to suppress iNOS in rodents and cell culture [12, 13]. In the present study, a short-term elevation of lipid levels was used to create insulin resistance. This was associated with reduced AMPKα2 activity in skeletal muscle and, as seen by others, increased skeletal muscle iNOS levels [46]. Infusion of Intralipid is sufficient to induce inflammatory markers in plasma [10] and skeletal muscle [47] of overnight fasted C57BL/6J mice. Indeed, Tnfα (also known as Tnf) expression increased in skeletal muscle after as little as 30 min of Intralipid [47]. Acute (24 h) infusion of TNFα suppresses AMPKα activity in mouse skeletal muscle [48]. Thus, it is quite possible that induction of TNFα over the 5 h Intralipid infusion protocol in the present study accounted for the observed suppression of AMPKα2 activity. However, the failure of AICAR-induced activation of AMPKα2 to normalise iNOS abundance or activity in skeletal muscle suggests that reduced AMPK activity is not directly linked to elevated iNOS activity. The dose of AICAR required to suppress iNOS in rodent skeletal muscle (bolus injection of 1,000 mg/kg [12]) is markedly higher than that used in the present study (constant infusion of 1 mg kg−1 min−1). Thus, as with our finding pertaining to muscle glucose uptake, it is possible that the ability of high-dose AICAR to suppress iNOS expression/activation occurs through a mechanism that is independent of AMPKα2.
In conclusion, the acute induction of insulin resistance in healthy mice via an Intralipid infusion is sufficient to reduce AMPKα2 activity and increase iNOS activation in skeletal muscle in vivo. This impaired AMPKα2 activity does not appear to contribute to the increase in iNOS activation or the impaired rates of insulin-stimulated muscle glucose uptake. However, AMPKα2 activity does appear to play a role in determining the cellular fate of glucose within skeletal muscle. Chronic insulin resistance is associated with reduced muscle AMPKα2 activity [6, 14] and impaired glycogen synthesis rates [49]. The findings of the present study suggest a link between defective AMPKα2 activation in skeletal muscle and reduced glycogen synthesis rates.
Abbreviations
- α2-KD:
-
Kinase-dead AMPKα2 subunit
- ACC:
-
Acetyl-CoA carboxylase
- AICAR:
-
5-Aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- AMPK:
-
AMP-activated protein kinase
- 2-[14C]DG:
-
2-Deoxy[14C]glucose
- EndoR a :
-
Endogenous rates of glucose appearance
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GIR:
-
Glucose infusion rate
- GS:
-
Glycogen synthase
- iNOS:
-
Inducible nitric oxide synthase
- R a :
-
Rates of glucose appearance
- R d :
-
Rates of glucose disappearance
- R g :
-
Tissue-specific indices of muscle glucose uptake
- ZMP:
-
5-Amino-4-imidazolecarboxamide ribotide
References
de Lange P, Moreno M, Silvestri E, Lombardi A, Goglia F, Lanni A (2007) Fuel economy in food-deprived skeletal muscle: signaling pathways and regulatory mechanisms. FASEB J 21:3431–3441
Seufert J (2006) A fixed-dose combination of pioglitazone and metformin: a promising alternative in metabolic control. Curr Med Res Opin 22(Suppl 2):S39–S48
Hardie DG (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8:774–785
Logie L, Harthill J, Patel K et al (2012) Cellular responses to the metal-binding properties of metformin. Diabetes 61:1423–1433
Iglesias MA, Ye JM, Frangioudakis G et al (2002) AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes 51:2886–2894
Lessard SJ, Chen Z-P, Watt MJ et al (2006) Chronic rosiglitazone treatment restores AMPKα2 activity in insulin-resistant rat skeletal muscle. Am J Physiol 290:E251–E257
Musi N, Hirshman MF, Nygren J et al (2002) Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51:2074–2081
Perreault M, Marette A (2001) Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 7:1138–1143
Torres SH, de Sanctis JB, de L Briceno M, Hernandez N, Finol HJ (2004) Inflammation and nitric oxide production in skeletal muscle of type 2 diabetic patients. J Endocrinol 181:419–427
Cha HN, Song SE, Kim YW, Kim JY, Won KC, Park SY (2011) Lack of inducible nitric oxide synthase prevents lipid-induced skeletal muscle insulin resistance without attenuating cytokine level. J Pharmacol Sci 117:77–86
Sugita H, Fujimoto M, Yasukawa T et al (2005) Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem 280:14203–14211
Pilon G, Dallaire P, Marette A (2004) Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem 279:20767–20774
Di Marco S, Mazroui R, Dallaire P et al (2005) NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol 25:6533–6545
Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM (2006) Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes 55:2277–2285
Mortensen B, Poulsen P, Wegner L et al (2009) Genetic and metabolic effects on skeletal muscle AMPK in young and older twins. Am J Physiol 297:E956–E964
Beck Jørgensen S, O’Neill H, Hewitt K, Kemp B, Steinberg G (2009) Reduced AMP-activated protein kinase activity in mouse skeletal muscle does not exacerbate the development of insulin resistance with obesity. Diabetologia 52:2395–2404
Sriwijitkamol A, Coletta DK, Wajcberg E et al (2007) Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes 56:836–848
Mu J, Barton ER, Birnbaum MJ (2003) Selective suppression of AMP-activated protein kinase in skeletal muscle: update on 'lazy mice'. Biochem Soc Trans 31:236–241
Ayala JE, Bracy DP, McGuinness OP, Wasserman DH (2006) Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55:390–397
Lee-Young RS, Griffee SR, Lynes SE et al (2009) Skeletal muscle AMP-activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem 284:23925–23934
Morgan CR, Lazarow A (1965) Immunoassay of pancreatic and plasma insulin following alloxan injection of rats. Diabetes 14:669–671
Ally A, Park G (1992) Rapid determination of creatine, phosphocreatine, purine bases and nucleotides (ATP, ADP, AMP, GTP, GDP) in heart biopsies by gradient ion-pair reversed-phase liquid chromatography. J Chromatogr 575:19–27
Fueger PT, Heikkinen S, Bracy DP et al (2003) Hexokinase II partial knockout impairs exercise-stimulated glucose uptake in oxidative muscles of mice. Am J Physiol 285:E958–E963
Chan TM, Exton JH (1976) A rapid method for the determination of glycogen content and radioactivity in small quantities of tissue or isolated hepatocytes. Anal Biochem 71:96–105
Golden S, Wals PA, Katz J (1977) An improved procedure for the assay of glycogen synthase and phosphorylase in rat liver homogenates. Anal Biochem 77:436–445
Steele R, Wall JS, DeBodo RC, Altszuler N (1956) Measurement of the size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol 187:15–24
Kraegen EW, James DE, Jenkins AB, Chisholm DJ (1985) Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol 248:E353–E362
Lee-Young RS, Ayala JE, Hunley CF et al (2010) Endothelial nitric oxide synthase is central to skeletal muscle metabolic regulation and enzymatic signaling during exercise in vivo. Am J Physiol 298:R1399–R1408
Bergeron R, Previs SF, Cline GW et al (2001) Effect of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes 50:1076–1082
Bergeron R, Russell RR 3rd, Young LH et al (1999) Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol 276:E938–E944
Boon H, Bosselaar M, Praet SF et al (2008) Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 51:1893–1900
Cuthbertson DJ, Babraj JA, Mustard KJ et al (2007) 5-Aminoimidazole-4-carboxamide 1-β-D-ribofuranoside acutely stimulates skeletal muscle 2-deoxyglucose uptake in healthy men. Diabetes 56:2078–2084
Shearer J, Fueger PT, Vorndick B et al (2004) AMP kinase-induced skeletal muscle glucose but not long-chain fatty acid uptake is dependent on nitric oxide. Diabetes 53:1429–1435
Stoppani J, Hildebrandt AL, Sakamoto K, Cameron-Smith D, Goodyear LJ, Neufer PD (2002) AMP-activated protein kinase activates transcription of the UCP3 and HKII genes in rat skeletal muscle. Am J Physiol Endocrinol Metab 283:E1239–E1248
Olsen GS, Hansen BF (2002) AMP kinase activation ameliorates insulin resistance induced by free fatty acids in rat skeletal muscle. Am J Physiol 283:E965–E970
Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF (2009) A-769662 activates AMPK β1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol 297:C1041–C1052
Aschenbach WG, Hirshman MF, Fujii N, Sakamoto K, Howlett KF, Goodyear LJ (2002) Effect of AICAR treatment on glycogen metabolism in skeletal muscle. Diabetes 51:567–573
Hunter RW, Treebak JT, Wojtaszewski JF, Sakamoto K (2011) Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60:766–774
Toole BJ, Cohen PT (2007) The skeletal muscle-specific glycogen-targeted protein phosphatase 1 plays a major role in the regulation of glycogen metabolism by adrenaline in vivo. Cell Signal 19:1044–1055
Kim YB, Shulman GI, Kahn BB (2002) Fatty acid infusion selectively impairs insulin action on Akt1 and protein kinase C lambda /zeta but not on glycogen synthase kinase-3. J Biol Chem 277:32915–32922
Barnes BR, Glund S, Long YC, Hjalm G, Andersson L, Zierath JR (2005) 5'-AMP-activated protein kinase regulates skeletal muscle glycogen content and ergogenics. FASEB J 19:773–779
Milan D, Jeon JT, Looft C et al (2000) A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288:1248–1251
Hoy AJ, Brandon AE, Turner N et al (2009) Lipid and insulin infusion-induced skeletal muscle insulin resistance is likely due to metabolic feedback and not changes in IRS-1, Akt, or AS160 phosphorylation. Am J Physiol 297:E67–E75
Viollet B, Andreelli F, Jorgensen SB et al (2003) The AMP-activated protein kinase α2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest 111:91–98
Jorgensen SB, Nielsen JN, Birk JB et al (2004) The α2-5'AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 53:3074–3081
Charbonneau A, Marette A (2010) Inducible nitric oxide synthase induction underlies lipid-induced hepatic insulin resistance in mice: potential role of tyrosine nitration of insulin signaling proteins. Diabetes 59:861–871
Kim BS, Cha HN, Kim YW, Kim JY, Dan JM, Park SY (2009) Inhibition of lipid infusion-induced skeletal muscle insulin resistance by cotreatment with tempol and glutathione in mice. J Pharmacol Sci 110:370–380
Steinberg GR, Michell BJ, van Denderen BJ et al (2006) Tumor necrosis factor α-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab 4:465–474
Del Prato S, Bonadonna RC, Bonora E et al (1993) Characterization of cellular defects of insulin action in type 2 (non-insulin-dependent) diabetes mellitus. J Clin Invest 91:484–494
Acknowledgements
We thank E.P. Donahue (Vanderbilt Mouse Metabolic Phenotyping Center Hormone and Analytical Core, Vanderbilt University, Nashville, TN, USA) for performing the HPLC analyses.
Funding
This work was supported by National Institutes of Health Grants DK054902 and DK059637 (to D.H. Wasserman). R.S. Lee-Young was supported by a mentor-based fellowship from the American Diabetes Association.
Contribution statement
RSLY, JSB, WHM, II, BAB, CMH and LK performed the experimental studies and analyses. RSLY and DHW designed the experimental studies and wrote, reviewed and edited the manuscript. CMH and LK reviewed and edited the manuscript and contributed to discussions. JSB, WHM, II and BAB reviewed the manuscript. All authors approved the final version of this manuscript.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee-Young, R.S., Bonner, J.S., Mayes, W.H. et al. AMP-activated protein kinase (AMPK)α2 plays a role in determining the cellular fate of glucose in insulin-resistant mouse skeletal muscle. Diabetologia 56, 608–617 (2013). https://doi.org/10.1007/s00125-012-2787-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2787-7