
Abstract
Aims/hypothesis
Irs2, which is upregulated by glucose, is important for beta cell plasticity. Cyclic AMP response element-binding protein (CREB) stimulates beta cell Irs2 expression and is a major calcium/calmodulin-dependent kinase (CaMK)IV target in neurons. We therefore hypothesised that CaMKIV mediates glucose-induced Irs2 expression in beta cells via CREB activation.
Methods
The functions of CaMKIV and CREB were investigated in MIN6 beta cells and mouse islets using the CaMK inhibitor KN62, the calcium chelator bapta-(AM) and the voltage-dependent calcium channel inhibitor nifedipine. Small interfering RNAs were used to silence endogenous CaMKIV production and expression vectors to overproduce constitutively active and dominant negative forms of CaMKIV and CREB. Irs1 and Irs2 expression were determined by quantitative PCR and Western blotting, and the role of CREB was also investigated by assessing its phosphorylation on serine 133.
Results
Increasing the glucose concentration from 2.5 to 25 mmol/l stimulated CREB phosphorylation on serine 133 and specifically stimulated Irs2 but not Irs1 expression. Similarly, overproduction of a constitutively active form of CaMKIV promoted sustained CREB phosphorylation and a significant increase in Irs2 but not Irs1 expression. In contrast, these stimulatory effects of glucose were all suppressed by overproducing an inactive CaMKIV mutant. Inhibition of glucose-induced calcium influx with nifedipine or chelation of intracellular calcium with bapta-(AM), as well as silencing of CaMKIV or inhibition of its activity with KN62 resulted in similar observations. Finally, overproduction of a dominant negative form of CREB completely suppressed glucose and CaMKIV stimulation of Irs2 expression.
Conclusions/interpretation
Our results suggest that the Ca2+/CaMKIV/CREB cascade plays a critical role in the regulation of Irs2 expression in beta cells.
Similar content being viewed by others


Introduction
Although it is generally accepted that type 2 diabetes mellitus is caused by a progressive decline in beta cell function [1] and beta cell mass [2, 3] following the development of insulin resistance [4, 5], several reports suggest that beta cell failure, rather than insulin resistance, is the primary defect, which occurs years before the onset of diabetes [1, 6]. This notion is supported by the general observation that type 2 diabetes mellitus does not develop in most obese individuals or in pregnant women, who can have severe insulin resistance [3, 7, 8], due to a compensatory process involving increased beta cell function and notably beta cell mass expansion [3, 9–11].
Short-term glucose administration promotes beta cell mass expansion [12–14], and recent observations implicate glucose-stimulated insulin secretion [15, 16] and Irs2 expression [17, 18] as the main upstream mechanisms. A strong correlation between IRS2 expression in beta cells with apoptosis, proliferation and type 2 diabetes suggests that IRS2 could become a target gene for future therapeutic intervention. Thus, Irs2 knockout mice exhibited a significant reduction in beta cell mass and developed the full phenotype of diabetes [19, 20], whereas targeted re-expression of Irs2 in beta cells increased their survival and promoted their growth through stimulation of proliferation [21]. More recently, it was reported that exendin-4, a stable glucagon-like peptide-1 (GLP-1) receptor agonist known to stimulate Irs2 expression and beta cell mass expansion, failed to do so in Irs2 knockout mice, thus linking the cAMP signalling pathway with Irs2 expression and activity [22].
The role of the cAMP/protein kinase A (PKA) pathway in glucose-regulated Irs2 expression and beta cell mass expansion was recently investigated and experiments using PKA inhibitors indicated that glucose-stimulated Irs2 expression was reduced by only 20% to 25% [17, 18]. These observations indicate that GLP-1 and glucose do not share the same signalling cascade to increase Irs2 expression, and that activation of the cAMP/PKA pathway is not the major mechanism by which glucose stimulates Irs2 expression.
Interestingly, calcium-dependent stimulation of dendritic growth in neurons is mediated by calcium/calmodulin-dependent kinase (CaMK)IV-induced cAMP response element-binding protein (CREB) activation, independently of cAMP/PKA stimulation [23]. Thus, since CamkIV (also known as Camk4) is expressed in beta cells [24], is activated by increases in intracellular Ca2+ levels as occurs following glucose metabolism [25] and CREB is known to stimulate Irs2 gene expression [26], it is possible that glucose regulates Irs2 levels in beta cells through a Ca2+/CaMKIV/CREB signalling cascade.
In the current study, the role of CaMKIV-induced CREB activation in mediating the glucose effects on Irs2 expression in MIN6 beta cells and mouse islets was examined through downregulation and overexpression studies that used small-interfering RNAs (siRNAs) and constitutively active or dominant negative forms of CaMKIV and CREB.
Methods
Ethics
Experiments involving animals were approved by the local ethics committee.
Cells, plasmids and reagents
MIN6 cells were a gift from Y. Oka and J.-I. Miyazaki (Third Department of Internal Medicine, Faculty of Medicine, University of Tokyo, Japan) and ICR mice were purchased from Harlan (Blackthorn, UK). Plasmids encoding the mouse constitutively active form of CaMKIV (ΔCaMKIV) and human kinase-dead form of CaMKIV (ΔK73ECaMKIV) were kindly provided by A. Ghosh (Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD, USA). pcDNA3.1 plasmids encoding the following forms of CREB were a gift from Professor D. D. Ginty (Department of Neuroscience, Howard Hughes Medical Institute, The Johns Hopkins University School of Medicine, Baltimore, USA): wild-type; constitutively active forms of CREB (DIEDML); and dominant negative forms of CREB (CREBm1). Metafectene Pro was from Biontex (Martinsried/Planegg, Germany). Glucose-free DMEM and FBS were purchased from Invitrogen (Paisley, UK). KN62 was from Calbiochem (Nottingham, UK). The mouse monoclonal anti-α-tubulin antibody, culture media, nifedipine, bapta-(AM), Histopaque-1077, collagenase (type XI), penicillin/streptomycin and l-glutamine were from Sigma Aldrich (Poole, UK). The rabbit polyclonal anti-IRS1 and anti-IRS2 antibodies were from Millipore (Watford, UK), the mouse monoclonal anti-CaMKIV antibody was from Clontech (Oxford, UK), and mouse monoclonal anti-CREB and anti-(133phosphoserine) CREB antibodies were from New England Biolabs (Hitchin, UK). The siRNA duplexes were obtained from Dharmacon (Cramlington, UK). The mRNA purification kit (RNeasy) was from Qiagen (Crawley, UK). Enhanced chemiluminescent kits for Western blotting were from GE Healthcare (Little Chalfont, UK). Horseradish peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were from Pierce Biotechnology (Rockford, IL, USA).
Cell culture, treatment and transfection
MIN6 beta cells were maintained in culture at 37°C in DMEM (25 mmol/l glucose) supplemented with 2 mmol/l glutamine, 10% (vol./vol.) FBS, 100 units/ml penicillin and 100 μg/ml streptomycin. To study the effect of glucose on Irs1 and Irs2 expression and on CREB phosphorylation, cells were maintained at 6 mmol/l glucose for 24 h followed by exposure to the experimental conditions described below (“Results”). Pre-treatment with the CaMK inhibitor KN62 (or DMSO vehicle, 45 min) and transient transfection with non-silencing RNA or siRNA duplexes designed to knock down CaMKIV levels were performed before final adjustment of the glucose concentration. High efficiency (~60–80%) transient transfection of MIN6 cells was achieved by electroporation (Nucleofector II; Amaxa, Cologne, Germany).
Mouse islets isolation, culture and siRNA transfection
Islets were isolated by collagenase digestion of mouse pancreas as described previously [27, 28] and maintained in culture for 24 to 48 h in RPMI 1640 medium (11 mmol/l glucose) supplemented with 10% (vol./vol.) FBS before use. To study the role of CaMKIV in primary tissue, mouse islets were transiently transfected with either a commercially available non-silencing RNA or with four siRNA duplexes designed to specifically reduce endogenous CaMKIV levels. The target sequences used were: 5′-gagauccucugggcgauuu-3′, 5′-ucaaggaaauauucgaaac-3′, 5′-ggugcuacauccauugugu-3′ and 5′-gggaugaagugucuuuaaa-3′. Transient transfection of mouse islets was performed using a two-step transfection protocol with Metafectene Pro as described [29].
Reverse transcription and quantitative polymerase chain reaction
Total RNA was isolated from ~5 × 105 MIN6 cells or ~150 mouse islets using RNeasy (Qiagen) according to the manufacturer’s instructions. Complementary DNAs were synthesised and quantitative PCR amplifications were performed as described previously [30]. Each sample value was normalised to beta-actin copy numbers. In all quantitative PCR experiments, the presence of possible contaminants was checked by control reactions in which amplification was performed in reaction mixtures without cDNA templates. Specificity of each primer pair was confirmed by melting curve analysis and agarose-gel electrophoresis of PCR products.
Western blot analysis
MIN6 cell protein extracts (50–75 μg) were separated on 10% (wt/vol.) polyacrylamide gels and transferred to polyvinylidene fluoride membranes, which were incubated for 16 h with antibodies directed against IRS1 (1:166 dilution), IRS2 (1:1,000 dilution), α-tubulin (1:2,000 dilution), CaMKIV (1:750 dilution), CREB (1:750 dilution) or phospho-CREB(Ser133) (1:750 dilution). After three washes in Tris buffered saline (pH 7.4) containing 0.05% Tween 20, the polyvinylidene fluoride membranes were incubated for another hour with horseradish peroxidase-coupled anti-rabbit or anti-mouse IgGs as appropriate (1:5,000 dilution). Binding of secondary antibodies was revealed by chemiluminescence.
Statistical analysis
Numerical data are expressed as means ± SEM. Differences between two groups were analysed by unpaired Student’s t test and considered significant at p < 0.05. Differences between several groups were analysed by one-way analysis of variance followed by Tukey’s honestly significant differences test.
Results
Glucose stimulates Irs2, but not Irs1 mRNA expression in beta cells
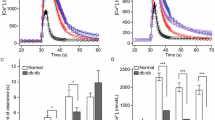
The effect of glucose on Irs1 and Irs2 mRNA expression was initially assessed using isolated mouse islets (Fig. 1a, b) or insulin-secreting MIN6 cells (Fig. 1c, d) that were maintained in culture for 24 h in the presence of 2.5 to 25 mmol/l glucose. As previously reported [17, 18], Irs2, but not Irs1 mRNA expression was significantly stimulated in a concentration-dependent manner by glucose in mouse islets and MIN6 cells, with a maximal effect occurring at 25 mmol/l.

Glucose regulates Irs2, but not Irs1 mRNA expression in beta cells. a, b Isolated mouse islets or (c, d) MIN6 beta cells were maintained in culture for 24 h in the presence of 6 mmol/l glucose and then exposed to 2.5 to 25 mmol/l glucose for another 24 h. Quantitative RT-PCR was used to determine mRNA expression of (a, c) Irs2 and (b, d) Irs1 relative to beta-actin. Values are per cent of those at 2.5 mmol/l glucose, shown as means ± SEM of three to five independent experiments. *p < 0.05 and **p < 0.01 vs 2.5 mmol/l glucose
Glucose-induced IRS2 production is calcium-dependent
As observed at the mRNA level, increasing the glucose concentration from 2.5 to 25 mmol/l also stimulated IRS2, but not IRS1 protein production in mouse islets (Fig. 2a, Electronic supplementary material [ESM] Fig. 1a) and MIN6 cells (Fig. 2b, ESM Fig. 1b). In addition, prevention of glucose-induced calcium influx and increase of intracellular calcium by addition of 10 μmol/l nifedipine or 10 μmol/l bapta-(AM) prior to glucose exposure completely suppressed this stimulatory effect on IRS2 production, but again without altering IRS1 protein levels. Taken together, these results are consistent with the notion that glucose specifically regulates IRS2 levels in a calcium-dependent manner.

Glucose-stimulated IRS2 upregulation is mediated by glucose-induced calcium influx. a Mouse islets and (b) MIN6 beta cells were maintained in culture for 24 h in the presence of 6 mmol/l glucose, pre-treated for 45 min with 10 μmol/l nifedipine (Nif), 10 μmol/l bapta-AM (Bapta) or DMSO vehicle (Con), and then exposed to 2.5 or 25 mmol/l glucose for another 24 h in the presence of the chemicals. Equivalent loading was confirmed by mouse monoclonal anti-α-tubulin antibody. Immunoblots are representative of three independent experiments
Glucose stimulation of Irs2 production is mediated by a CaMK pathway
The data in Figs 1 and 2 indicate that glucose stimulates Irs2 mRNA expression and IRS2 protein levels in a concentration-dependent manner in MIN6 beta cells and in mouse islets. We therefore used MIN6 beta cells to further examine the molecular mechanisms downstream of calcium influx, which mediate this effect. Thus, knowing that CREB has the ability to induce beta cell Irs2, but not Irs1 expression [26], and that CaMKI and CaMKIV mediate the calcium effect on CREB activation in the GH3 growth hormone-secreting cell line [31], we tested the hypothesis that glucose-stimulated Irs2 expression in beta cells is regulated by a CaMK pathway. This was achieved by assessing the effect of the non-selective CaMK inhibitor KN62 (30 μmol/l) on glucose-stimulated CREB phosphorylation at serine 133 and on IRS2 protein levels.
As shown in Fig. 3a and ESM Fig. 2a, a low level of CREB phosphorylation at serine 133 could be detected at 6 mmol/l glucose (t = 0 min). Challenging the MIN6 cells with 12 mmol/l glucose resulted in a rapid, but transient increase of CREB phosphorylation, with a maximal effect observed after 5 min of exposure. In addition, exposure of MIN6 cells to 12 and 25 mmol/l glucose for 24 h resulted in enhanced IRS2 protein production (Fig. 3b, ESM Fig. 2b), consistent with data displayed in Fig. 2. However, the presence of KN62 suppressed the stimulatory effects of glucose on CREB phosphorylation (Fig. 3a, ESM Fig. 2a) and greatly reduced the glucose-dependent upregulation of IRS2 (Fig. 3b, ESM Fig. 2b). These observations are consistent with the notion that glucose-stimulated Irs2 expression is mediated, at least in part, by a CaMK–CREB signalling cascade. In addition, the specific involvement of this signalling cascade in regulation of Irs2 expression by glucose was demonstrated by our observations that neither increased glucose concentrations nor the use of KN62 altered Irs1 expression levels (Figs 2 and 3c, ESM Fig. 2c).

Glucose stimulates IRS2 production via a CaMK. a Immunoblot showing the role of CaMKs in glucose-induced CREB activation as illustrated by detection of CREB phosphorylation (pCREB) at serine 133. MIN6 cells were pre-incubated with 6 mmol/l glucose for 24 h, treated with the CaMK inhibitor KN62 (30 μmol/l) or DMSO for 45 min, and then exposed to 12 mmol/l glucose for additional times as indicated (0 to 30 min). Levels of pCREB were monitored by Western blotting using total CREB as loading controls. b Immunoblot showing that glucose stimulates IRS2, but not (c) IRS1 protein production in a CaMK-dependent manner in MIN6 beta cells that were treated with 2.5, 12 and 25 mmol/l glucose for 24 h in the presence or absence of 30 μmol/l KN62. Equivalent loading was confirmed by mouse monoclonal anti-α-tubulin antibody. All immunoblots are representative of three independent experiments
Glucose-stimulated Irs2 expression is controlled by CaMKIV
CaMKI, CaMKII and CaMKIV all have the ability to stimulate CREB phosphorylation at serine 133, but a recent study in astrocytes suggests that only CaMKIV has the capacity to mediate the calcium-dependent activation of this transcription factor [32]. Thus, to investigate whether the CaMKIV isoform also mediates glucose-stimulated Irs2 expression in beta cells, MIN6 cells stably expressing ΔCaMKIV or ΔK75ECaMKIV were generated and their expression profiles characterised by Western blotting. It can be seen from Fig. 4a that native MIN6 cells produced only the 60 kDa native CaMKIV (lane 1), whereas cells overproducing the dominant negative and constitutively active CaMKIV mutants also produced truncated 37 kDa (lane 2) and 35 kDa (lane 3) CaMKIV proteins. As expected, overproduction of ΔCaMKIV was associated with sustained CREB phosphorylation (Fig. 4b, ESM Fig. 3), whereas overproduction of ΔK75ECaMKIV resulted in an almost complete loss of glucose-induced CREB phosphorylation (Fig. 4b, ESM Fig. 3). To further investigate whether these mutant-induced variations in CREB phosphorylation levels were also associated with a similar alteration in Irs2 mRNA expression, we compared the effects of low (2.5 mmol/l), stimulatory (12 mmol/l) and supra-physiological (25 mmol/l) glucose concentrations. As shown in Fig. 5a, stable overproduction of ΔCaMKIV significantly upregulated Irs2 mRNA expression at 2.5 mmol/l and 12 mmol/l glucose, but did not modify it at 25 mmol/l glucose. In contrast, overproduction of ΔK75ECaMKIV reduced Irs2 mRNA expression at all glucose concentrations (Fig. 5c). The specificity of these effects on Irs2 was confirmed by observations that overproduction of these two CaMKIV mutants did not modify Irs1 expression patterns at any glucose concentrations tested (Fig. 5b, d).

Glucose-induced CREB phosphorylation at serine 133 is mediated by CaMKIV. a Stable transfection of ΔCaMKIV and ΔK75ECaMKIV in MIN6 beta cells. Sub-confluent MIN6 cells were co-transfected with pcDNA3.1/ΔCaMKIV or pcDNA3.1/ΔK75ECaMKIV expression vectors or with pcDNA3.1 alone, and pcDNA3.1-positive colonies selected by maintenance in the presence of 800 μg/ml G418. Single colonies were selected, expanded in culture and tested for ΔCaMKIV and ΔK75ECaMKIV by Western blotting using a monoclonal anti-CaMKIV antibody. The immunoblot shows the abundance of endogenous CaMKIV (~60 kDa), of constitutively active ΔCaMKIV (~35 kDa) and of dominant negative ΔK75ECaMKIV (~37 kDa) in extracts of MIN6 cells stably expressing pcDNA3.1 (lane 1), pcDNA3.1/ΔK75ECaMKIV (lane 2) or pcDNA3.1/ΔCaMKIV (lane 3). b CREB phosphorylation (pCREB) at serine 133 in response to ΔCaMKIV or ΔK75ECaMKIV overabundance in MIN6 cells. MIN6 cells stably transfected with pcDNA3.1, pcDNA3.1/ΔCaMKIV or pcDNA3.1/ΔK75ECaMKIV as labelled were maintained in culture in the presence of 6 mmol/l glucose for 24 h before being challenged with 12 mmol/l glucose for times as indicated (0 to 30 min). Total CREB production was used as loading control. Immunoblots shown are representative of three separate experiments

CaMKIV mediates the stimulatory glucose effect on Irs2 mRNA expression. a, b pcDNA3.1 (Con), pcDNA3.1/ΔCaMKIV (Δ) and (c, d) pcDNA3.1/ΔK75ECaMKIV (ΔK75E) transfectant MIN6 beta cells were exposed to glucose as indicated for 24 h and quantitative RT-PCR was used to determine mRNA expression of (a, c) Irs2 and (b, d) Irs1 relative to beta-actin. Values are per cent of those at 2.5 mmol/l glucose (Con), shown as means ± SEM of three independent experiments. *p < 0.05 and **p < 0.01
CaMKIV mediates glucose-induced Irs2 expression via CREB activation
The data displayed in Fig. 6a show that similar results were obtained at the protein level, with glucose increasing IRS2 production in control cells. They also show that stable overproduction of ΔCaMKIV resulted in a significant increase in IRS2 protein at 2.5 mmol/l glucose, with no further increase when glucose concentrations were increased to 12 and 25 mmol/l. In contrast, competitive inhibition of endogenous CaMKIV following stable expression of ΔK75ECaMKIV reduced IRS2 levels at 2.5 mmol/l glucose and also decreased glucose-induced IRS2 production.

The glucose/CaMKIV signalling cascade uses CREB activation to stimulate IRS2 production. a pcDNA3.1 (control), pcDNA3.1/ΔCaMKIV and pcDNA3.1/ΔK75ECaMKIV transfectant MIN6 cells were maintained in culture with 6 mmol/l glucose for 24 h, then exposed to glucose as indicated for another 24 h before IRS2 protein levels were determined by Western blotting. Equivalent loading was confirmed by a mouse monoclonal anti-α-tubulin antibody. The immunoblot is representative of three independent experiments. b, c MIN6 cells were transiently transfected with pcDNA3.1 (Control, Con) or various expression vectors coding for ΔCaMKIV, ΔK75ECaMKIV, wild-type CREB, CREBm1 or CREBDIEDML. At 24 h after transfection, the cells were treated with glucose as indicated for an additional 24 h before determination of IRS2 protein levels by Western blotting. Equivalent loading was confirmed with mouse monoclonal anti-α-tubulin antibody. Each immunoblot is representative of three independent experiments. Values are per cent of those at 2.5 and 6 mmol/l glucose (b, c) respectively, shown as means ± SEM of optical density ratios from three independent experiments. *p < 0.05 and **p < 0.01; †† p < 0.01
To establish whether a direct link between glucose, CaMKIV, CREB and IRS2 production exists in beta cells, CREBm1 and the constitutively active form of CREB (CREBDIEDML) were transiently overexpressed in MIN6 cells and the resulting effects on glucose- and CaMKIV-induced stimulation of IRS2 protein levels were analysed by Western blotting. As shown in Fig. 6b, IRS2 production was stimulated in a glucose- and CaMKIV-dependent manner, confirming the results obtained in our stable transfection studies (Fig. 6a). The data also show that excess levels of CREBm1, as with excess ΔK75ECaMKIV, suppressed the stimulatory effect of high glucose concentrations (12 and 25 mmol/l) on IRS2 production. As expected, Fig. 6c shows that overproduction of CREBDIEDML at a non-stimulatory glucose concentration of 6 mmol/l resulted in a marked increase in IRS2 protein in MIN6 cells. In addition, whereas simultaneous excess of ΔCaMKIV and the wild-type form of CREB generated an additive effect on IRS2 production, excess levels of CREBm1 completely abolished the stimulatory effect of ΔCaMKIV.
Reduction of CaMKIV levels decreases glucose-induced Irs2 expression
To confirm that the data obtained in our transfection studies were not the consequence of non-specific effects of ΔCaMKIV or ΔK75ECaMKIV overproduction, we used siRNA duplexes to knock down CaMKIV levels in native MIN6 beta cells. In these experiments, a marked reduction in CaMKIV protein production was obtained after 24 h exposure to siRNAs and sustained for at least 48 h (Fig. 7a). Interestingly, increasing the glucose concentration from 2.5 to 25 mmol/l for 24 h not only increased Irs2 mRNA levels (Fig. 7b) as already observed (Figs 1 and 5), but also resulted in significant stimulation of CamkIV mRNA expression (Fig. 7c), suggesting a correlation between CaMKIV expression/activity and Irs2 expression. Consistent with this inter-relationship, knock-down of CaMKIV levels with siRNA duplexes decreased Irs2 mRNA expression at 25 mmol/l glucose to a level not significantly different from that observed at 2.5 mmol/l glucose (Fig. 7b) and also significantly reduced the stimulatory effect of 25 mmol/l glucose on CamkIV mRNA expression (Fig. 7c). The specificity of these effects was demonstrated by our observation that the use of these siRNAs did not alter Irs1 mRNA expression (Fig. 7d). In addition, these siRNAs did not reduce CamkIIa (also known as Camk2a) mRNA expression 24 h after transfection of MIN6 beta cells (Fig. 7e), suggesting that CaMKII expression and function were also not altered in our experimental conditions.

siRNA-mediated reduction of CaMKIV levels suppresses glucose-induced Irs2 mRNA expression in MIN6 beta cells. a Immunoblot showing the effect of four siRNA duplexes designed to reduce CaMKIV protein levels in MIN6 cells that were maintained in culture for 24 or 48 h in the presence of 25 mmol/l glucose. b–e MIN6 cells were transfected either with a non-interfering siRNA duplex (Con) or four duplexes designed to silence CaMKIV production, and exposed to glucose as indicated for 24 h. Quantitative RT-PCR was used to determine mRNA expression of Irs2 (b), CamkIV (c), Irs1 (d) and CamkIIa (e) relative to those of beta-actin. Values are per cent of those at 2.5 mmol/l glucose, shown as means ± SEM of three to four independent experiments. *p < 0.05 relative to Con mRNA determined at 2.5 mmol/l glucose; † p < 0.05 and †† p < 0.01 relative to Con mRNA determined at 25 mmol/l glucose
To extend the observations we made in islets that glucose stimulates Irs2 upregulation in a calcium-dependent manner (Figs 1 and 2) and to determine whether this occurs via activation of the CaMKIV–CREB pathway as it does in MIN6 beta cells, several key experiments were performed using freshly isolated mouse islets. It can be seen from Fig. 8a–d that exposing islets to 25 mmol/l glucose for 24 h resulted in a significant stimulation of CamkIV mRNA expression and that this effect was associated with a significant increase in Irs2 mRNA expression. In contrast, 25 mmol/l glucose failed to upregulate Irs2 mRNA expression in mouse islets that had been transfected with CamkIV siRNA duplexes, which reduced CamkIV mRNA expression (Fig. 8a), but not CamkIIa (Fig. 8d) to a level not significantly different from that observed in the presence of 2.5 mmol/l glucose. As shown in Fig. 8e and ESM Fig. 4a–c, increasing the glucose concentration from 2.5 to 25 mmol/l also stimulated CaMKIV and IRS2 production at the protein level in mouse islets, while downregulation of CaMKIV levels by siRNAs entirely suppressed these effects. Figure 8 and ESM Fig. 4 also demonstrate that Irs1 mRNA expression and protein levels were not modified when the glucose concentration was increased from 2.5 to 25 mmol/l or when the islets were transfected with the CamkIV siRNA duplexes. Finally, the observation that CREB phosphorylation at serine 133 was significantly increased in mouse islets in response to 25 mmol/l glucose (Fig. 8d, ESM Fig. 4d) confirms our results in MIN6 cells (Figs 3 and 4) and further supports the hypothesis that glucose stimulates Irs2 expression via activation of a CaMKIV–CREB signalling cascade in beta cells.

siRNA-mediated reduction in CaMKIV levels suppresses glucose-induced Irs2 mRNA expression and protein levels in mouse islets. a–e Mouse islets were transfected either with a non-interfering siRNA duplex (Con) or four duplexes designed to silence CaMKIV production, and maintained in culture for 24 h in the presence of 5.5 mmol/l glucose, then exposed to glucose as indicated for another 24 h. Quantitative RT-PCR was used to determine mRNA expression of (a) CamkIV, (b) Irs2, (c) Irs1 and (d) CamkIIa relative to those of beta-actin. Values are a percentage of those at 2.5 mmol/l glucose, shown as means ± SEM of three independent experiments. *p < 0.05 and **p < 0.01 relative to mRNA determined at 2.5 mmol/l glucose; † p < 0.05 and †† p < 0.01 relative to mRNA determined at 25 mmol/l glucose. e Mouse islets were treated as above and CaMKIV, IRS2 and IRS1 protein levels were analysed by Western blotting. Equivalent loading was confirmed by mouse monoclonal anti-α-tubulin antibody. f Immunoblot showing the stimulatory effect of glucose on CREB phosphorylation (pCREB) at serine 133 in mouse islets. Mouse islets were maintained in culture for 24 h in the presence of 2.5 mmol/l glucose and then exposed to 25 mmol/l glucose for another 7 min. Immunoblots are representative of three independent experiments
Discussion
CaMKIV is a multifunctional enzyme whose function is best understood in neurons, where it inhibits apoptosis and stimulates growth in a calcium- and CREB-dependent manner [23, 33–37]. CamkIV is also expressed by pancreatic beta cells [24], but its roles in beta cells have not been fully defined. Earlier reports that glucose and GLP-1 receptor agonists regulate beta cell mass through CREB- and IRS2-dependent inhibition of apoptosis and stimulation of proliferation [17, 18, 21, 26, 38–41] led us to examine the role of the CaMKIV–CREB cascade in the regulation of Irs2 expression by beta cells.
Our results demonstrate that glucose stimulates Irs2 expression in islets and MIN6 cells in a calcium-dependent manner and provide evidence for the first time that this is mediated by the CaMKIV–CREB pathway. Indeed, glucose-dependent Irs2 mRNA and protein upregulation were substantially reduced when either ΔK75ECaMKIV or CREBm1 were overabundant in MIN6 cells or following siRNA-induced reduction of endogenous CaMKIV content. In contrast, excess levels of the constitutively active forms of CaMKIV (ΔCaMKIV) or of CREB (CREBdiedml) resulted in enhanced Irs2 expression, while the stimulatory effect of ΔCaMKIV was suppressed by coproduction of the dominant negative CREB mutant (CREBm1 ). These novel observations in MIN6 cells are consistent with previous in vitro [23, 42] and in vivo [33] reports in other tissues, demonstrating that CaMKIV controls CREB transcriptional activity. The observation that only Irs2, but not Irs1 expression levels were modified when glucose concentration was increased or the constitutively active form of CaMKIV was overproduced also confirms previous observations of selective upregulation of Irs2 by glucose [17, 18], as well as demonstrating the specificity of this glucose–CaMKIV effect. We also observed that stable ΔCaMKIV production by MIN6 cells produced significant elevations of Irs2 mRNA at 2.5 and 12 mmol/l glucose compared with native MIN6 cells, but this did not occur at 25 mmol/l glucose. These observations suggest that production of the constitutively active ΔCaMKIV in MIN6 cells by-passed the requirement for glucose-stimulated calcium entry and enabled the cells to maximally stimulate Irs2 expression independently of a glucose stimulus.
However, whereas glucose-induced CREB phosphorylation at serine 133 was abolished by the calcium/calmodulin kinase inhibitor KN62 or in MIN6 cells stably transfected with ΔK75ECaMKIV (Figs. 3a and 4b), glucose-stimulated IRS2 protein production was only partially reduced (Figs 3b and 6a). This suggests that part of the stimulatory effect of glucose on IRS2 abundance may be independent of CaMKIV and that alternative mechanisms of action exist. One possible alternative signalling cascade might involve glucose-induced increases in intracellular cAMP levels [43], which have been shown to promote MIN6 cell CREB phosphorylation at serine 133 and CREB activation, but with a delayed time course compared with depolarising stimuli [26]. This hypothesis is supported by previous observations showing that H-89 and KT5720, two PKA inhibitors, reduced glucose-stimulated Irs2 mRNA expression and protein levels by approximately 25% in rat islets [18].
MIN6 beta cells were used for many of the experiments presented in this study because they have several of the key functional characteristics of primary beta cells [44] and are readily amenable to stable transfection. However, MIN6 cells are a transformed beta cell line expressing the SV40 large T-antigen, which keeps them in a proliferative state, and they are also adapted to maintenance in media containing high glucose concentrations. Therefore, to ensure that the data obtained using MIN6 cells did not reflect signalling cascades present in cell lines but not in primary beta cells, key experiments were repeated using isolated mouse islets. We found that glucose stimulated a calcium-dependent upregulation of Irs2 in islets, with stimulatory profiles similar to those seen in MIN6 cells; glucose also stimulated CREB phosphorylation. In addition, our experiments in mouse islets, in which glucose-stimulated Irs2 upregulation was lost when CaMKIV production was transiently knocked down, confirmed the existence, in mouse islets, of the glucose/CaMKIV/Irs2 cascade that we had identified in MIN6 cells. Taken together, our data imply that CaMKIV plays a central role in the regulation of Irs2 expression in islets. Previous studies using the HIT-T15 and INS-1 insulin-secreting cell lines suggest that CaMKIV also mediates glucose-induced insulin gene expression and insulin secretion [24, 25], and that it stimulates glucokinase expression [44]. In addition, CamkIV is a target gene for the canonical wingless-type MMTV integration site family (WNT)/β-catenin signalling pathway [45], whose signalling-associated transcription factor TCF7L2 is a diabetes susceptibility gene [46, 47] known to regulate beta cell proliferation [48], insulin gene expression and insulin secretion [49]. Thus, a central role for CamKIV in islet function implicates it as a useful target gene for the development of future drug therapies to treat type 2 diabetes mellitus.
The results presented here establish a critical role for CaMKIV in Irs2 expression. This, together with previous observations showing a reduction in beta cell mass due to increased beta cell apoptosis in Irs2 knockout mice [19–21], suggests that CaMKIV might regulate beta cell survival and proliferation. Preliminary data, obtained in our laboratory and demonstrating that overexpression of the constitutively active form of CaMKIV in MIN6 cells stimulates proliferation and reduces caspase-3/7 activities, are consistent with this hypothesis (D. S. Muller, S. J. Persaud, B. Liu and P. M Jones, unpublished data). The precise role of Irs2 in these CaMKIV-mediated effects is now being investigated.
In conclusion, the current study demonstrates for the first time that CaMKIV has a central role in CREB-dependent mechanisms by which glucose regulates Irs2 expression in beta cells. Moreover, since Irs2 deficiency has been linked with the progressive development of type 2 diabetes mellitus, our results suggest that finding a mechanism to stimulate CamKIV expression and/or activity could have a significant clinical impact in the future for patients with type 2 diabetes mellitus.
Abbreviations
- CaMK:
-
Calcium/calmodulin-dependent kinase
- ΔCaMKIV :
-
Constitutively active form of CaMKIV
- CREB:
-
cAMP response element-binding protein
- CREBDIEDML :
-
Constitutively active form of CREB
- CREBm1 :
-
Dominant negative form of CREB
- GLP-1:
-
Glucagon-like peptide-1
- ΔK75ECaMKIV :
-
Dominant negative form of CaMKIV
- PKA:
-
Protein kinase A
- siRNA:
-
Small-interfering RNA
References
Kahn SE (2001) Beta cell failure: causes and consequences. Int J Clin Pract 123:13–18
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta cell deficit and increased beta cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Deng S, Vatamaniuk M, Huang X et al (2004) Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes 53:624–632
Muoio DM, Newgard CB (2008) Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9:193–205
Kahn SE, Carr DB, Faulenbach MV, Utzschneider KM (2008) An examination of beta cell function measures and their potential use for estimating beta cell mass. Diabetes Obes Metab 4:63–76
Chiasson JL, Rabasa-Lhoret R (2004) Prevention of type 2 diabetes: insulin resistance and beta cell function. Diabetes 53:S34–S38
Polonsky KS (2000) Dynamics of insulin secretion in obesity and diabetes. Int J Obes Relat Metab Disord 2:S29–S31
Buchanan (2001) Pancreatic B-cell defects in gestational diabetes: implications for the pathogenesis and prevention of type 2 diabetes. J Clin Endocrinol Metab 86:989–993
Butler AE, Janson J, Soeller WC, Butler PC (2003) Increased beta cell apoptosis prevents adaptive increase in beta cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52:2304–2314
Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE (2005) Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 48:1350–1358
Parsons JA, Brelje TC, Sorenson RL (1992) Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 130:1459–1466
Bonner-Weir S, Deery D, Leahy JL, Weir GC (1989) Compensatory growth of pancreatic beta cells in adult rats after short-term glucose infusion. Diabetes 38:49–53
Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A (2003) Specific and combined effects of insulin and glucose on functional pancreatic beta cell mass in vivo in adult rats. Endocrinology 144:2717–2727
Teil GM, Trivedi N, Jonas JC et al (2001) Adaptation of beta cell mass to substrate oversupply: enhanced function with normal gene expression. Am J Physiol Endocrinol Metab 280:E788–E796
Muller D, Jones PM, Persaud SJ (2006) Autocrine anti-apoptotic and proliferative effects of insulin in pancreatic beta cells. FEBS Lett 580:6977–6980
Aikin R, Hanley S, Maysinger D et al (2006) Autocrine insulin action activates Akt and increases survival of isolated human islets. Diabetologia 49:2900–2909
Amacker-Francoys I, Mohanty S, Niessen M, Spinas GA, Trub T (2005) The metabolisable hexoses d-glucose and d-mannose enhance the expression of IRS-2 but not of IRS-1 in pancreatic beta cells. Exp Clin Endocrinol Diabetes 113:423–429
Lingohr MK, Briaud I, Dickson LM et al (2006) Specific regulation of IRS-2 expression by glucose in rat primary pancreatic islet beta cells. J Biol Chem 281:15884–15892
Kubota N, Tobe K, Terauchi Y et al (2000) Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta cell hyperplasia. Diabetes 49:1880–1889
Withers DJ, Gutierrez JS, Towery H et al (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900–904
Hennige AM, Burks DJ, Ozcan U et al (2003) Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. J Clin Invest 112:1521–1532
Park S, Dong X, Fisher TL et al (2006) Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J Biol Chem 281:1159–1168
Redmond L, Kashani AH, Ghosh A (2002) Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34:999–1010
Yu X, Murao K, Sayo Y et al (2004) The role of calcium/calmodulin-dependent protein kinase cascade in glucose upregulation of insulin gene expression. Diabetes 53:1475–1481
Ban N, Yamada Y, Someya Y et al (2000) Activating transcription factor-2 is a positive regulator in CaM kinase IV-induced human insulin gene expression. Diabetes 49:1142–1148
Jhala US, Canettieri G, Screaton RA et al (2003) cAMP promotes pancreatic beta cell survival via CREB-mediated induction of IRS2. Genes Dev 17:1575–1580
King A, Lock J, Xu G, Bonner-Weir S, Weir GC (2005) Islet transplantation outcomes in mice are better with fresh islets and exendin-4 treatment. Diabetologia 48:2074–2079
Papadimitriou A, King AJ, Jones PM, Persaud SJ (2007) Anti-apoptotic effects of arachidonic acid and prostaglandin E2 in pancreatic beta cells. Cell Physiol Biochem 20:607–616
Muller DS, Jones PM, Persaud SJ (2007) Expression of a potent anti-apoptotic protein in MIN6 b-cells using METAFECTENE PRO. Available from www.biontex.com/con_4_6_4/cms/upload/pdf/Muller_MP_en.pdf. Accessed 4 January 2011
Muller D, Huang GC, Amiel S, Jones PM, Persaud SJ (2006) Identification of insulin signaling elements in human beta cells: autocrine regulation of insulin gene expression. Diabetes 55:2835–2842
Sun P, Lou L, Maurer RA (1996) Regulation of activating transcription factor 1 and the cAMP response element-binding protein by Ca2+/calmodulin-dependent protein kinases type I, II, and IV. J Biol Chem 271:3066–3073
Murray PD, Kingsbury TJ, Krueger BK (2009) Failure of Ca(2+)-activated, CREB-dependent transcription in astrocytes. Glia 57:828–834
Ho N, Liauw JA, Blaeser F et al (2000) Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20:6459–6472
Hansen MR, Bok J, Devaiah AK, Zha XM, Green SH (2003) Ca2+/calmodulin-dependent protein kinases II and IV both promote survival but differ in their effects on axon growth in spiral ganglion neurons. J Neurosci Res 72:169–184
Yano S, Tokumitsu H, Soderling TR (1998) Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396:584–587
Sée V, Boutillier AL, Bito H, Loeffler JP (2001) Calcium/calmodulin-dependent protein kinase type IV (CaMKIV) inhibits apoptosis induced by potassium deprivation in cerebellar granule neurons. FASEB J 15:134–144
Deisseroth K, Heist EK, Tsien RW (1998) Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392:198–202
Jansson D, Ng ACH, Fu A, Depatie C, Al Azzabi M, Screaton RA (2008) Glucose controls CREB activity in islet cells via regulated phosphorylation of TORC2. Proc Natl Acad Sci U S A 105:10161–10166
Pechhold K, Koczwara K, Zhu X et al (2009) Blood glucose levels regulate pancreatic beta cell proliferation during experimentally-induced and spontaneous autoimmune diabetes in mice. PLos ONE 4:e4827
Bonner-Weir S (1994) Regulation of pancreatic beta cell mass in vivo. Recent Prog Horm Res 49:91–104
Hoorens A, Van de CM, Kloppel G, Pipeleers D (1996) Glucose promotes survival of rat pancreatic beta cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest 98:1568–1574
Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR (1994) Characterization of Ca2+/calmodulin-dependent protein kinase IV: role in transcriptional regulation. J Biol Chem 269:15520–15527
Landa LR Jr, Harbeck M, Kaihara K et al (2005) Interplay of Ca2+ and cAMP signalling in the insulin-secreting MIN6 β-cell line. J Biol Chem 280:31294–31302
Ishihara H, Asano T, Tsukuda K et al (1993) Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia 36:1139–1145
Arrázola MS, Varela-Nallar L, Colombres M et al (2009) Calcium/calmodulin-dependent protein kinase type IV is a target gene of the Wnt/beta-catenin signaling pathway. J Cell Physiol 221:658–667
Jin T, Liu L (2008) The Wnt signalling pathway effector TCF7L2 and type 2 diabetes mellitus. Mol Endocrinol 22:2383–2392
Jin T (2008) The Wnt signalling pathway and diabetes mellitus. Diabetologia 51:1771–1780
Liu Z, Habener JF (2008) Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J Biol Chem 283:8723–8735
Loder MK, da Silva XG, McDonald A, Rutter GA (2008) TCF7L2 controls insulin gene expression and insulin secretion in mature pancreatic beta cells. Biochem Soc Trans 36:357–359
Acknowledgements
We are grateful to J. I. Miyazaki (University of Osaka, Osaka, Japan) for the provision of MIN6 cells, to A. Gosh (University of California, San Diego, CA, USA) for CaMKIV plasmids, to D. D. Ginty (John Hopkins University, Baltimore) for CREB plasmids, to P. Marsh (King’s College London, London, UK) for helping with plasmid amplifications, and to J. Bowe and A. King (King’s College London, London, UK) for assisting with mouse islet isolation. We gratefully acknowledge The Eli Lilly International Foundation for grant support. B. Liu was supported by an Overseas Research Students Postgraduate Award. D. S. Muller was a Diabetes UK RD Lawrence Fellow.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Quantitative evaluation of the immunoblots showing that glucose-stimulated IRS2 upregulation is mediated by glucose-induced calcium influx. As described in Fig. 2, mouse islets (a, b) and MIN6 beta cells (c, d) were maintained in culture for 24 h in the presence of 6 mmol/l glucose, pre-treated for 45 min with 10 μmol/l nifedipine (Nif), 10 μmol/l bapta-AM (Bapta) or DMSO (vehicle, Con), and then exposed to glucose as indicated for another 24 h in the presence of the chemicals. Bars show means ± SEM of three to five independent experiments. **p < 0.01 relative to IRS2 or IRS1 levels determined at 2.5 mmol/l glucose; † p < 0.05 and †† p < 0.01 relative to IRS2 or IRS1 levels determined at 25 mmol/l glucose (PDF 236 kb)
ESM Fig. 2
Quantitative evaluation of the immunoblots showing that glucose stimulates IRS2 production via a CaMK. a As described in Fig. 3, MIN6 cells were pre-incubated with 6 mmol/l glucose for 24 h, treated with the CaMK inhibitor KN62 (30 μmol/l) or DMSO for 45 min, and then exposed to 12 mmol/l glucose for an additional 1 to 30 min. Bars show means of phosphorylated CREB (pCREB):CREB ratios ± SEM of three independent experiments. *p < 0.05 and **p < 0.01 relative to pCREB/CREB ratios determined at time 0. b, c Effect of glucose as indicated on IRS2 and IRS1 protein levels. Bars show means ± SEM of three independent experiments. *p < 0.05 and **p < 0.01 relative to IRS2 or IRS1 levels determined at 2.5 mmol/l glucose (PDF 209 kb)
ESM Fig. 3
Quantitative evaluation of the immunoblots showing that glucose-induced CREB phosphorylation at serine 133 is mediated by CaMKIV. As described in Fig. 4b, MIN6 cells stably transfected with pcDNA3.1, pcDNA3.1/ΔCaMKIV or pcDNA3.1/ΔK75ECaMKIV were maintained in culture in the presence of 6 mmol/l glucose for 24 h before being challenged with 12 mmol/l glucose for 1 to 30 min. CREB levels and phosphorylation levels at serine 133 were then quantified by optic densitometry. Bars show means of phosphoryltated CREB (pCREB)/CREB ratios ± SEM of three independent experiments. *p < 0.05 and **p < 0.01 relative to pCREB/CREB ratios determined at time 0 in the control groups (PDF 163 kb)
ESM Fig. 4
Quantitative evaluation of the immunoblots showing that siRNA-mediated reduction in CaMKIV levels suppresses glucose-induced IRS2 production in mouse islets. a–c As described in Fig. 8, mouse islets were transfected either with a non-interfering siRNA duplex (Con) or four duplexes designed to silence CaMKIV production, and maintained in culture for 24 h in the presence of 5.5 mmol/l glucose. Islets were then exposed to glucose as indicated for another 24 h. Bars show means ± SEM of three independent experiments. *p < 0.05 and **p < 0.01 relative to IRS2, CaMKIV or IRS1 levels determined at 2.5 mmol/l glucose; †† p < 0.01 relative to IRS2, CaMKIV or IRS1 levels determined at 25 mmol/l glucose. d Mouse islets were maintained in culture for 24 h in the presence of 2.5 mmol/l glucose, and exposed or not to 25 mmol/l glucose for another 7 min. Total CREB and phosphorylated CREB (pCREB) levels were determined by optic densitometry. Bars show means of pCREB/CREB ratios ± SEM of three independent experiments. **p < 0.01 relative to pCREB/CREB ratios determined at 2.5 mmol/l glucose. (PDF 248 kb)
Rights and permissions
About this article
Cite this article
Persaud, S.J., Liu, B., Sampaio, H.B. et al. Calcium/calmodulin-dependent kinase IV controls glucose-induced Irs2 expression in mouse beta cells via activation of cAMP response element-binding protein. Diabetologia 54, 1109–1120 (2011). https://doi.org/10.1007/s00125-011-2050-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2050-7