
Abstract
The recent major increase in the global incidence of type 2 diabetes suggests that most cases of this disease are caused by changes in environment and lifestyle. All major risk factors for type 2 diabetes (overnutrition, low dietary fibre, sedentary lifestyle, sleep deprivation and depression) have been found to induce local or systemic low-grade inflammation that is usually transient or milder in individuals not at risk for type 2 diabetes. By contrast, inflammatory responses to lifestyle factors are more pronounced and prolonged in individuals at risk of type 2 diabetes and appear to occur also in the pancreatic islets. Chronic low-grade inflammation will eventually lead to overt diabetes if counter-regulatory circuits to inflammation and metabolic stress are compromised because of a genetic and/or epigenetic predisposition. Hence, it is not the lifestyle change per se but a deficient counter-regulatory response in predisposed individuals which is crucial to disease pathogenesis. Novel approaches of intervention may target these deficient defence mechanisms.
Similar content being viewed by others


Introduction
According to estimates of the World Health Organization and the International Diabetes Federation, the prevalence of diabetes has increased from 100–135 million affected adults worldwide in 1994–1995 to approximately 246 million in 2007, with more than 95% of cases considered to be type 2 diabetes. Longer periods of observation are available from single countries such as Germany, where the diabetes prevalence (types 1 and 2) has risen more than tenfold from 0.6% in 1960 to 6.9% in 2005 [1].
Since the changes in the incidence of type 2 diabetes have occurred too rapidly to reflect genetic causes, the vast majority of new cases of type 2 diabetes are likely to be caused by changes in lifestyle and/or environment during the last decades. Candidate epigenetic factors are the consumption of excess energy, in particular from increases in the intake of saturated fatty acids, sugar-sweetened beverages and starchy food, and the consumption of less fibre, in conjunction with a more sedentary lifestyle, i.e. less muscle work—all factors promoting obesity. Psychosocial factors may also have contributed to the steep increase in the prevalence type 2 diabetes, in particular increases in the prevalence of short duration of sleep and depressive mood [2, 3]. A general diabetes risk factor with increasing prevalence is age. Other known risk factors for diabetes, such as smoking and infections, may have contributed little, because of only small changes in exposure in many areas of the world.
In this review we expand the concept of an immune or inflammatory origin of type 2 diabetes that has been discussed previously [4]. We discuss whether the changes of lifestyle in past decades have promoted inflammatory processes, thereby causing increased insulin demands and beta cell death.
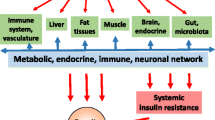
Lifestyle factors conferring risk for type 2 diabetes may elicit many different cellular responses, e.g. endoplasmic reticulum stress, mitochondrial dysfunction, excess and ectopic fat storage, innate immune-receptor activation and insulin resistance. Considered from a higher organisational level, the key tissues affected are the vessels, the immune system, adipose tissue, skeletal muscle, liver, brain, gut with microbiota, and the pancreatic islets. Cellular responses in any of these organs have been suggested in the past to play a primary role in the pathogenesis of type 2 diabetes because they may lead to an imbalance between insulin need and insulin production. We will argue that a local and/or systemic inflammatory reaction occurs in response to virtually all diabetes-promoting lifestyle factors which have been analysed in that regard. We propose that a critical feature in the development of type 2 diabetes is the inability to sufficiently contain such usually transient inflammatory reactions by regulatory feedback mechanisms, and that this is an important determinant of whether lifestyle factors promote type 2 diabetes development.

Glucose/starch
There is ample evidence that in vitro exposure of endothelial or mononuclear cells to high glucose concentrations (usually ≥10 mmol/l) induces the expression of inflammation-associated genes, such as those coding for IL-6, IL-8, monocyte chemoattractant protein-1 and several adhesion molecules [5]. One straightforward mechanistic explanation is that there is more substrate for mitochondrial activity as a consequence of increased glucose influx, and that increased respiratory activity gives rise to enhanced release of superoxide anions [6]. However, other studies suggest that glucose-induced superoxide formation stems from glucose-6-phosphate dehydrogenase and phosphokinase C-dependent activation of NAD(P)H oxidase [7]. Subsequently, the production of pro-inflammatory immune mediators occurs via the activation of mitogen-activated kinases p38 and Jun N-terminal kinase (JNK), other tyrosine kinases, transcription factors such as transcription factor activator protein-1 (AP-1) and nuclear factor κB (NFκB), and/or poly(ADP-ribose) polymerase [8, 9].
In keeping with these in vitro observations, a dietary pattern with high consumption of sugar-sweetened beverages, sweets and white bread is associated with increased levels of circulating pro-inflammatory markers such as C-reactive protein (CRP) [10]. The concomitant low intake of cereal fibre, fruit and vegetables may be involved, along with other diet-associated lifestyle factors. In general, intervention trials report results similar to those from observational studies, i.e. a diet low in sugar and starch but high in fruit and vegetables leads to lower than average levels of C-reactive protein [11]. Unfortunately, such trials cannot provide information on effects of single nutrients.
The acute effects of glucose or starch consumption are seen during a 1–4 h period following consumption. Challenge with 75 g glucose was found to more than double spontaneous oxygen radical production from leucocytes 2–3 h later [12]. Increased activation of NFκB was found in response to glucose or a bread meal [13], and there was an increase in circulating levels of IL-6 and TNF-α during a hyperglycaemic clamp [14]. In some studies, glucose consumption led to lower levels of IL-6 or TNF-α [15]. It is noteworthy that patients with impaired glucose tolerance or type 2 diabetes appear to exhibit a more pronounced pro-inflammatory response to dietary glucose at the level of circulating IL-6 and TNF-α [14], or at the level of IL6 and TNFα (also known as TNF) gene expression in leucocytes, both in vivo and in vitro [16]. This suggests that early in the progression towards type 2 diabetes the ability to contain pro-inflammatory responses to glucose or starchy meals is compromised. In this regard, type 1 diabetes may be different, because a pattern of systemic inflammatory markers similar to that in type 2 diabetes is not observed, at least in the absence of chronic complications. However, the immune disease process per se is associated with systemic upregulation of several cytokines and chemokines, and residual beta cell function appears positively associated with levels of anti-inflammatory mediators [17].
There is increasing evidence that the induction of immune mediators is an important component of glucose-induced beta cell apoptosis. High glucose upregulates the production of apoptosis-stimulating fragment in human beta cells, with concomitant expression of IL1 mRNA and production of protein. Indeed, glucose-induced human beta cell apoptosis could be blocked by the IL-1 receptor antagonist (IL-1RA) [18].
As described above for the systemic response to glucose challenge, there seems to be an impaired anti-inflammatory feedback circuit in islets, i.e. deficient production of antagonistic IL-1RA in type 2 diabetic islets was found [19]. Concomitantly, there is increased IL-1ß production in such islets [20].

Saturated fatty acids
At concentrations around postprandial peak plasma levels, palmitate (200–500 µmol/l) induced oxygen radical production and the release of pro-inflammatory cytokines from several cell types after 1 or more days in vitro [21, 22]. The molecular pathways involved include: mitochondrial superoxide formation; protein kinase C activation, with subsequent induction of NAD(P)H oxidase; induction of diacylglycerol synthesis; and the induction of endoplasmic reticulum stress [21, 23, 24]. All of these pathways may directly cause deficient insulin signalling, but they also lead to the activation of pro-inflammatory gene transcription through the activation of NFκB, AP-1 or cAMP response element binding protein [22, 24].
A sizeable pro-inflammatory response is also seen in the hours following a meal rich in saturated fat, and includes increases in: the levels of markers of oxidative stress; the production of superoxide, IL-1, IL-6, IL-18, adhesion molecules and NAD(P)H oxidase; the numbers of leucocytes; and activation of NFκB [25, 26]. Infusion of a triacylglycerol solution for 4 h similarly caused NFκB activation, oxygen radical formation from blood cells and increased levels of circulating macrophage-migration inhibitory factor, a potent pro-inflammatory cytokine. These responses occurred in parallel with a rise of non-esterified fatty acid concentrations in plasma [27]. After lipid infusion, the concentration of ceramides also increases in plasma, causing activation of NFκB [28].
Co-ingestion of oils rich in mono- or polyunsaturated fatty acids dampened postmeal inflammatory responses [29]. Postmeal inflammation was more pronounced or prolonged in persons with obesity, metabolic syndrome or type 2 diabetes, or was only observed in such persons compared with lean and healthy individuals [30–32].
As yet not understood is the variation in results from studies into the types and kinetics of postmeal production of cytokines. Some studies found transient decreases in serum cytokine levels, others a lack of a protective effect of unsaturated fatty acids [33]. This may be due in part to the opposing effects of n-3 and n-6 polyunsaturated fatty acids. [34], but the evidence in favour of this concept is limited.
An additional pathway promoting low-grade inflammation is the leakage of endotoxin from the colon upon ingestion of a high-fat meal, which appears to be relevant for the induction of hepatic insulin resistance [35].
Currently, the strongest evidence in favour of a pathogenic role of the pro-inflammatory response to dietary fat comes from animal models. Here, interaction of non-esterified saturated fatty acids with the lipopolysaccharide (LPS) receptor Toll-like receptor (TLR)4 and subsequent activation of NFκB appears to be the major signalling pathway in the induction of a pro-inflammatory response and concomitant insulin resistance [36, 37]. Of particular importance is the finding that TLR4 dysfunction prevents dietary-fat-induced insulin resistance in adipose tissue, muscle and liver, and often there is less adiposity and slowed hepatic steatosis despite adipocyte hypertrophy [37, 38]. Suppression of the canonical activation of NFκB by disruption of the gene encoding inhibitor of κB (IκB) kinase beta also prevented high-fat-diet-induced insulin resistance; a lack of this enzyme in myeloid cells is sufficient for this effect to occur [39]. Conversely, constitutively active IκB kinase beta in hepatocytes causes profound hepatic and moderate systemic insulin resistance [40].
Postmeal lipaemia may directly affect beta cell survival, a phenomenon termed lipotoxicity. Fatty acids potentiate IL-1-mediated beta cell toxicity [41]. Fatty acids, but not high glucose, markedly reduce human islet peroxisome proliferator-activated receptor (PPAR)γ production [42], and since PPARγ agonists have anti-inflammatory properties and protect islets from cytokine toxicity [43], fatty acids may reduce beta cell anti-inflammatory defence mechanisms. Of note, fatty acids have not been reported to activate intra-islet innate immunity. Both inflammatory cytokines and saturated fatty acids cause beta cell apoptosis via the intrinsic pathway, whereas saturated fatty acids also activate an NFκB- and nitric-oxide-independent endoplasmic reticulum stress death pathway [43–45].

Dietary fibre and other food components
In the Finnish Diabetes Prevention Study increased fibre intake was one of two independent factors predicting decreases in circulating levels of CRP and IL-6 [46]. A recent review of dietary intervention trials reported that in six out of seven trials, daily consumption of 3–8 g fibre per megajoule led to significant decrements in plasma CRP concentrations of 25–54% [47].
As with saturated fat, information on the pathophysiological relevance of the immunomodulatory property of dietary fibre is only available from animal models. Feeding oligofructose to leptin-deficient (ob/ob) mice decreased intestinal permeability and systemic levels of pro-inflammatory cytokines as well as visceral and subcutaneous adiposity, and increased portal plasma levels of glucagon-like peptide-2 [48].
Taken together, the limited data available are consistent with a role of dampened inflammation as a mechanism of action of fibre as a factor for diabetes protection.
Other food components
There is a multitude of other food components which are known to modulate immune reactivity and have been suggested to promote the development of type 2 diabetes. Prominent examples are adducts of glucose to protein or lipids as a consequence of thermal processing of food [49]. Deficiencies of vitamin D and calcium have also been linked with type 2 diabetes [50]. It is not known whether exposure to these risk factors has increased along with the global rise in diabetes incidence during the last decades.

Excess nutrients/obesity
Obesity is a key risk factor for type 2 diabetes. A prominent characteristic of diabetes-promoting lifestyle change is weight gain. Since the first reports of local inflammation in obese adipose tissue in 2003 [51, 52] it has become clear that fat tissue with hypertrophic adipocytes often gives rise to the influx and activation of immune cells, mostly macrophages but also of T cells [53]. This is more pronounced in visceral, hepatic and epicardial than in subcutaneous fat [54]. Concomitantly, there is death of single adipocytes and secretion of pro-inflammatory cytokines, chemokines and other adipokines from immune cells and/or adipocytes [55] that contributes to further recruitment of macrophages and adipositis. Other associated changes are ectopic fat deposition in several organs, including beta cells, endoplasmic reticulum stress, compromised fatty acid oxidation, increased levels of circulating non-esterified fatty acids and other lipids [24] and as observed in mice, dysfunction of neural regulatory systems such as leptin resistance, again closely linked with local inflammation [56]. The intimate relationship between visceral obesity and inflammation is underscored by findings that weight reduction causes downregulation of inflammation in visceral fat as well as in the circulation [57, 58]. Despite differences in visceral obesity among sex and race, visceral fat remains associated with systemic markers of inflammation among the populations studied [59]. However, the degree of inflammation may differ with ethnicity [60].
In the presence of central obesity the increase in serum concentration of immune mediators is usually less than twofold, whereas during infection it is tenfold or more. Such a modest increase may not be sufficient to be of pathological relevance, particularly when considering that the spread of serum concentrations between individuals is usually 10- to 100-fold [61]. A different situation emerges when paracrine effects of immune mediators are being considered. Paracrine concentrations of immune mediators are usually close to or reach the nanomolar range for activated macrophages [62] and this is sufficient to induce insulin resistance. Indeed, co-culture of adipocytes with macrophages causes impairment of insulin signalling [63]. In addition to paracrine effects it is conceivable that the functions of liver cells are affected by immune mediators released from visceral adipose tissue because of the vicinity of the circulating blood. In favour of a preferentially paracrine effect of locally produced immune mediators is the observation in mice that blockade of pro-inflammatory signalling via IκB kinase beta in hepatocytes only prevented high-fat-diet-induced insulin resistance in the liver, and not systemically [39].
The different pathways of immune-mediator-induced insulin resistance have been reviewed in detail previously and will not be reviewed again here. In essence, there may be direct action in the target cell interfering with insulin signalling, mostly by causing the phosphorylation of insulin receptor substrate 1 at serine residues, and there may be indirect action by modulating the secretion of mediators from fat tissue, liver and other organs that have an impact on insulin resistance, such as adiponectin [64].
Overnutrition and obesity increase insulin requirements, impose metabolic stress on beta cells and promote cellular exhaustion, but the mechanisms of action are poorly understood. Beta cell insulin resistance impairs trophic, proliferative and survival signals, e.g. the phosphatidylinositol-3 kinase–phosphokinase B pathway, and promotes early beta cell failure in an animal model [65]. Increased beta cell secretory activity sensitises beta cells to the pro-apoptotic action of inflammatory mediators, mainly when increasing substrate metabolism [66], but these effects are difficult to isolate from the effects of substrate-induced islet innate immunity [67]. It appears that such detrimental consequences are only clinically relevant if beta cell function and/or defences are compromised a priori, genetically, because of unfavourable conditions during organ development [68].

Muscle work
The contracting skeletal muscle secretes a number of immune mediators, such as IL-6, IL-8 and IL-15, and this can also be demonstrated in human skeletal muscle cells after mechanical strain in vitro [69]. The IL-6 serum concentrations increase up to 100-fold so that systemic effects are possible, e.g. the promotion of insulin resistance and lipolysis, whereas in skeletal muscle insulin action is augmented [70]. It seems difficult to reconcile this systemic pro-inflammatory effect of intense muscle work with the observed anti-inflammatory effect of regular exercise [46, 71]. One probable explanation is that the acute pro-inflammatory bout following intense physical exercise is followed by the increase in systemic levels of the anti-inflammatory cytokine IL-10 [72]. At least some of the IL-10 may come from the muscle itself, and indeed mice lacking IL-10 exhibit exaggerated production of IL-6 in skeletal muscle [73].

Psychosocial factors
Psychosocial lifestyle changes in past decades probably include sleep curtailment, because of voluntary bedtime restriction in modern societies. Sleep loss is a risk factor for insulin resistance and diabetes, and experimental studies in healthy volunteers have shown that sleep restriction causes decreased insulin sensitivity and glucose tolerance [2]. This may be due to the concomitant upregulation of systemic inflammation, as demonstrated by increased circulating concentrations of CRP or pro-inflammatory cytokines [74]. Pro-inflammatory changes were observed in peripheral blood mononuclear cells, such as activation of NFκB [75].
Another psychosocial factor that contributes to recent lifestyle changes is depression. Although major depression may not be a clearly defined entity, depressive symptoms have been found to be associated with type 2 diabetes in a bidirectional manner. A meta-analysis of nine trials concluded that depressed adults have a 37% higher risk of developing type 2 diabetes [3]. Furthermore, adults with diabetes exhibit a higher risk of depression [76], as do children with metabolic syndrome [77]. Depression has also been found to be strongly associated with increased levels of the circulating pro-inflammatory markers CRP, IL-6 and IL-1 [78]. Depressive symptoms and deterioration of insulin sensitivity are known to occur in response to treating chronic hepatitis C with interferon γ and the former appears to be due to the activation of an inflammatory response in the brain that interacts with serotonin metabolism [79]. The cytokines IL-1, IL-6 and TNF-α appear to cause general effects on cognitive function by impairing synaptic plasticity, neurogenesis and neuromodulation [80]. Twin studies found common genetic control of depressive symptoms and inflammatory markers [81]. Pharmacological antidepressive treatment with selective serotonin-reuptake inhibitors or tricyclic antidepressants was found to lower levels of TNF-α and CRP [82].

Lessons from immune intervention trials
The concept of an inflammatory cause of type 2 diabetes has led to attempts to halt disease progression by anti-inflammatory intervention. In a placebo-controlled study 70 patients with long-standing type 2 diabetes and poor metabolic control self-administered recombinant interleukin-1 receptor antagonist by daily subcutaneous injection for 13 weeks [83]. There was a reduction of fasting glucose levels within 1 week and significant reductions in HbA1c after 4 and 13 weeks (from 8.69% to 8.37% vs 8.23% to 8.37% in the control group), improved beta cell function and reduction in inflammatory markers but no change in insulin sensitivity, NEFA or glucose oxidation, adipokines or muscle gene expression [83, 84]. The effects on beta cell function and inflammatory markers were sustained after 39 weeks [85]. Interestingly, the baseline endogenous circulating IL-1RA level was markedly lower in patients responding to IL-1RA therapy, and this difference was stable at 52 weeks of follow-up. Subsequent analysis demonstrated that a common polymorphism (allelic frequency in the general population of 43%) of the gene encoding IL-1RA (IL1RN) accounted for the deficiency in circulating IL-1RA. In this respect this polymorphism, which occurs in a significant proportion of type 2 diabetic patients, is part of a group of diseases of IL-1RA deficiency that also includes rare mutations in IL1RN that lead to severe multisystem inflammatory syndromes that are highly responsive to IL-1RA treatment [86].
No clear effects on insulin sensitivity or beta cell function were observed after administration of recombinant TNF-α inhibitors despite a decrease in systemic levels of pro-inflammatory immune mediators [87], except for some cases of concomitant hypoglycaemia [88]. The doses of inhibitors administered were effective in rheumatoid arthritis but whether they were sufficient to suppress the paracrine effects of IL-1 or TNF-α in fat or muscle tissue is not known.
Treatment modalities that aim at reversing many of the lifestyle effects described above exert beneficial effects on metabolic control, beta cell function (where measured) and inflammatory markers in parallel. These include weight reduction [58] and dietary changes plus increased physical activity [46] or bariatric surgery [89].
The glucose-lowering potency of salicylates has been known for more than a century and experimental studies have suggested that this effect may be due to their anti-inflammatory action, primarily via inhibition of IκB kinase beta. The first clinical trials with high dose (≥3 g) salsalate or the salicylate derivative trifusal indeed showed that fasting glucose levels decreased in non-diabetic, mostly obese, persons while insulin concentrations increased in the context of reduced insulin clearance [90–92]. In a pilot trial of 7 g aspirin/day for 2 weeks in nine patients with type 2 diabetes, similar changes were observed, with mean fasting plasma glucose levels falling from 9.1 to 6.9 mmol/l [93].

Synthesis and open questions/suggestions for studies
We suggest here that lifestyle factors confer risk for type 2 diabetes through essentially the same mechanism, i.e. by inducing a chronic pro-inflammatory state in the innate immune system at a systemic level, in pancreatic islets and in obese adipose tissue. The pathways by which these factors induce activation of innate immunity are diverse and include increased production of superoxide from mitochondria and NAD(P)H oxidase, endoplasmic reticulum stress, release of LPS from the colon into the circulation, and intracellular accumulation of pro-inflammatory lipid metabolites, all of which may result in the activation of protein kinase C, mitogen-activated protein kinases or NFκB signalling, as has been reviewed elsewhere [24, 35, 67, 94].
A most important finding in understanding the relationship between innate immunity and diabetes development is that persons with type 2 diabetes, metabolic syndrome or obesity exhibit more pronounced and longer-lasting inflammatory responses to a challenge than metabolically normal controls in most studies [14, 30–32]. Pro-inflammatory reactions may even be observed in persons with metabolic syndrome only [16]. This suggests that the inflammatory response to diabetes-promoting lifestyle factors is usually self-limiting. With increased exposure to pro-inflammatory challenges, the counter-regulatory anti-inflammatory feedback inhibition mechanism may start to fail and chronic inflammation ensue in those genetically predisposed (Fig. 1). The resulting inflammatory stress and decreased insulin sensitivity are expected to promote beta cell failure in those individuals whose beta cell function and defence or regeneration capacity is compromised. It is of interest that a deficiency of counter-regulatory immune mechanism has been described to be associated with ageing, a major diabetes risk factor. Both innate and adaptive immunity are affected and a key observation is prolonged activation state of NFκB. The resulting chronic low-grade state of inflammation is discussed as a mechanism underlying ageing [95].

Lifestyle factors cause insulin resistance and beta cell failure via systemic and local low-grade inflammation. Inflammatory and metabolic stress will only become detrimental to the organism if there is a deficient ability to cope with these insults. CREB, cAMP-response-element binding protein
Examples of this concept of deficient anti-inflammatory feedback inhibition mechanisms are the intra-islet imbalance in IL-1 agonism and antagonism detailed above, and TLR4 involved in signalling pathways leading to inflammation in response to diabetes-promoting factors. First, TLR4 has been identified as one signalling receptor mediating the pro-inflammatory effects of saturated fatty acids; animals lacking functional TLR4 are resistant to fat-induced insulin resistance [36]. Second, TLR4 is the primary receptor on Kupffer cells for LPS leaking from the gut into circulation, which occurs in response to a high-fat diet [35]. Third, TLR4 has also been found to be critical in the delivery of pro-inflammatory signals of high glucose concentrations, although it is improbable that glucose directly binds to TLR4 [96]. Direct binding of palmitate to TLR4 could also not be demonstrated [97]. Thus, high glucose or saturated fat stress probably gives rise to the production of endogenous ligands for TLR4. Interestingly, tissue damage in response to ischaemia–reperfusion also requires intact TRL4, again indicating that endogenous TLR4 ligands are produced during local stress [98]. We have previously identified heat shock protein 60 as one such ‘danger antigen’ [99].
Without counter-regulatory feedback, stimulation of TLR4 might result in a sustained chronic inflammatory process. The self-limiting circuit is evident from the kinetics of cytokine production following exposure of macrophages to LPS. Within a few hours there is a strong pro-inflammatory response with massive secretion of numerous pro-inflammatory cytokines, including TNF-α, IL-1ß, IL-6 and IL-8. However, this is followed by the production of anti-inflammatory cytokines, notably IL-10 and IL-1RA during the following hours. As a consequence, macrophages become refractory to re-stimulation by LPS for this period. This state of suppressed LPS responsiveness is called LPS tolerance [62]. Hence, TLR4 induces pro-inflammatory and anti-inflammatory responses. Another potent pathway of limiting inflammation is the induction of anti-inflammatory mediators. These mechanisms appear to fail in chronic inflammation conditions.
The above described alternative pro- and anti-inflammatory states of macrophages have led to the concept of M1 and M2 macrophages. The anti-inflammatory state cannot only be reached as a late response to pro-inflammatory stimuli but also directly by anti-inflammatory stimuli, e.g. exposure to IL-4, IL-13 or IL-10 [62]. Infusions of lipids in mice treated with IL-10 did not induce insulin resistance [100].
We therefore propose that prodiabetic lifestyle factors become pathogenic if anti-inflammatory counter-regulatory mechanisms fail. Although this may be genetically controlled, any genes involved are yet to be identified. Examples of such regulatory circuits are the IL-1/IL-1RA balance and those associated with TLR4 signalling. One consequence would be a deficiency in LPS tolerance in type 2 diabetes. However, as discussed, chronic low-grade inflammation and concomitantly increased insulin needs will not necessarily result in dysglycaemia unless beta cells are limited in their capacity to cope with these conditions.
Further elucidation of the mechanism of anti-inflammatory feedback inhibitory circuits and why they fail during the development of type 2 diabetes seems warranted. A logical approach for preventing type 2 diabetes in persons at risk would be to aim at re-establishing mechanisms underlying the self-limiting of inflammatory responses to prodiabetic lifestyle factors.
Abbreviations
- AP-1:
-
Transcription factor activator protein-1
- CRP:
-
C-reactive protein
- IκB:
-
Inhibitor of κB
- IL-1RA:
-
IL-1 receptor antagonist
- JNK:
-
Jun N-terminal kinase
- LPS:
-
Lipopolysaccharide
- NFκB:
-
Nuclear factor κB
- PPAR:
-
Peroxisome proliferator-activated receptor
- TLR:
-
Toll-like receptor
References
Hauner H (2007) Diabetesepidemie und Dunkelziffer. In: Deutsche Diabetes Union (ed) Deutscher Gesundheitsbericht Diabetes. Kirchheim, Mainz, pp 7–12
Tasali E, Leproult R, Spiegel K (2009) Reduced sleep duration or quality: relationships with insulin resistance and type 2 diabetes. Prog Cardiovasc Dis 51:381–391
Knol MJ, Twisk JW, Beekman AT, Heine RJ, Snoek FJ, Pouwer F (2006) Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 49:837–845
Kolb H, Mandrup-Poulsen T (2005) An immune origin of type 2 diabetes? Diabetologia 48:1038–1050
Haubner F, Lehle K, Munzel D, Schmid C, Birnbaum DE, Preuner JG (2007) Hyperglycemia increases the levels of vascular cellular adhesion molecule-1 and monocyte-chemoattractant-protein-1 in the diabetic endothelial cell. Biochem Biophys Res Commun 360:560–565
El Osta A, Brasacchio D, Yao D et al (2008) Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med 205:2409–2417
Dasu MR, Devaraj S, Jialal I (2007) High glucose induces IL-1beta expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab 293:E337–E346
Devaraj S, Venugopal SK, Singh U, Jialal I (2005) Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-α and -β. Diabetes 54:85–91
Piconi L, Quagliaro L, Da Ros R et al (2004) Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: the role of poly(ADP-ribose) polymerase. J Thromb Haemost 2:1453–1459
Fung TT, McCullough ML, Newby PK et al (2005) Diet-quality scores and plasma concentrations of markers of inflammation and endothelial dysfunction. Am J Clin Nutr 82:163–173
Esposito K, Marfella R, Ciotola M et al (2004) Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA 292:1440–1446
Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P (2000) Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab 85:2970–2973
Dickinson S, Hancock DP, Petocz P, Ceriello A, Brand-Miller J (2008) High-glycemic index carbohydrate increases nuclear factor-kappaB activation in mononuclear cells of young, lean healthy subjects. Am J Clin Nutr 87:1188–1193
Esposito K, Nappo F, Marfella R et al (2002) Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation 106:2067–2072
Manning PJ, Sutherland WH, Hendry G, de Jong SA, McGrath M, Williams SM (2004) Changes in circulating postprandial proinflammatory cytokine concentrations in diet-controlled type 2 diabetes and the effect of ingested fat. Diabetes Care 27:2509–2511
Kempf K, Rose B, Herder C et al (2007) The metabolic syndrome sensitizes leukocytes for glucose-induced immune gene expression. J Mol Med 85:389–396
Pfleger C, Mortensen HB, Hansen L et al (2008) Association of IL-1ra and adiponectin with C-peptide and remission in patients with type 1 diabetes. Diabetes 57:929–937
Maedler K, Sergeev P, Ris F et al (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Maedler K, Sergeev P, Ehses JA et al (2004) Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci USA 101:8138–8143
Boni-Schnetzler M, Thorne J, Parnaud G et al (2008) Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93:4065–4074
Lambertucci RH, Hirabara SM, Silveira LR, Levada-Pires AC, Curi R, Pithon-Curi TC (2008) Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J Cell Physiol 216:796–804
Laine PS, Schwartz EA, Wang Y et al (2007) Palmitic acid induces IP-10 expression in human macrophages via NF-kappaB activation. Biochem Biophys Res Commun 358:150–155
Yu HY, Inoguchi T, Kakimoto M et al (2001) Saturated non-esterified fatty acids stimulate de novo diacylglycerol synthesis and protein kinase c activity in cultured aortic smooth muscle cells. Diabetologia 44:614–620
Schenk S, Saberi M, Olefsky JM (2008) Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest 118:2992–3002
Aljada A, Mohanty P, Ghanim H et al (2004) Increase in intranuclear nuclear factor kappaB and decrease in inhibitor kappaB in mononuclear cells after a mixed meal: evidence for a proinflammatory effect. Am J Clin Nutr 79:682–690
Esposito K, Nappo F, Giugliano F et al (2003) Meal modulation of circulating interleukin 18 and adiponectin concentrations in healthy subjects and in patients with type 2 diabetes mellitus. Am J Clin Nutr 78:1135–1140
Tripathy D, Mohanty P, Dhindsa S et al (2003) Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 52:2882–2887
Haus JM, Kashyap SR, Kasumov T et al (2009) Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58:337–343
Pacheco YM, Lopez S, Bermudez B, Abia R, Villar J, Muriana FJ (2008) A meal rich in oleic acid beneficially modulates postprandial sICAM-1 and sVCAM-1 in normotensive and hypertensive hypertriglyceridemic subjects. J Nutr Biochem 19:200–205
Blackburn P, Despres JP, Lamarche B et al (2006) Postprandial variations of plasma inflammatory markers in abdominally obese men. Obesity (Silver Spring) 14:1747–1754
Nappo F, Esposito K, Cioffi M et al (2002) Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients: role of fat and carbohydrate meals. J Am Coll Cardiol 39:1145–1150
Patel C, Ghanim H, Ravishankar S et al (2007) Prolonged reactive oxygen species generation and nuclear factor-kappaB activation after a high-fat, high-carbohydrate meal in the obese. J Clin Endocrinol Metab 92:4476–4479
Poppitt SD, Keogh GF, Lithander FE et al (2008) Postprandial response of adiponectin, interleukin-6, tumor necrosis factor-alpha, and C-reactive protein to a high-fat dietary load. Nutrition 24:322–329
Schmitz G, Ecker J (2008) The opposing effects of n-3 and n-6 fatty acids. Prog Lipid Res 47:147–155
Cani PD, Neyrinck AM, Fava F et al (2007) Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50:2374–2383
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015–3025
Milanski M, Degasperi G, Coope A et al (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29:359–370
Poggi M, Bastelica D, Gual P et al (2007) C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50:1267–1276
Arkan MC, Hevener AL, Greten FR et al (2005) IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11:191–198
Cai D, Yuan M, Frantz DF et al (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11:183–190
Aarnes M, Schonberg S, Grill V (2002) Fatty acids potentiate interleukin-1beta toxicity in the beta-cell line INS-1E. Biochem Biophys Res Commun 296:189–193
Lupi R, del Guerra S, Marselli L et al (2004) Rosiglitazone prevents the impairment of human islet function induced by fatty acids: evidence for a role of PPARgamma2 in the modulation of insulin secretion. Am J Physiol Endocrinol Metab 286:E560–E567
Kim EK, Kwon KB, Koo BS et al (2007) Activation of peroxisome proliferator-activated receptor gamma protects pancreatic beta-cells from cytokine-induced cytotoxicity via NF kappaB pathway. Int J Biochem Cell Biol 39:1260–1275
Grunnet LG, Aikin R, Tonnesen MF T et al (2009) Pro-inflammatory cytokines activate the intrinsic apoptotic pathway in β-cells. Diabetes 58:1807–1815
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY (2001) Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50:69–76
Herder C, Peltonen M, Koenig W et al (2009) Anti-inflammatory effect of lifestyle changes in the Finnish Diabetes Prevention Study. Diabetologia 52:433–442
North CJ, Venter CS, Jerling JC (2009) The effects of dietary fibre on C-reactive protein, an inflammation marker predicting cardiovascular disease. Eur J Clin Nutr 63:921–933
Cani PD, Possemiers S, van de Wiele T et al (2009) Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58:1091–1103
Uribarri J, Cai W, Sandu O, Peppa M, Goldberg T, Vlassara H (2005) Diet-derived advanced glycation end products are major contributors to the body’s AGE pool and induce inflammation in healthy subjects. Ann N Y Acad Sci 1043:461–466
Holick MF (2008) Diabetes and the vitamin D connection. Curr Diab Rep 8:393–398
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Kintscher U, Hartge M, Hess K et al (2008) T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 28:1304–1310
Hamdy O, Porramatikul S, Al Ozairi E (2006) Metabolic obesity: the paradox between visceral and subcutaneous fat. Curr Diabetes Rev 2:367–373
Strissel KJ, Stancheva Z, Miyoshi H et al (2007) Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56:2910–2918
Velloso LA, Araujo EP, de Souza CT (2008) Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation 15:189–193
Clement K, Viguerie N, Poitou C et al (2004) Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J 18:1657–1669
Puglisi MJ, Fernandez ML (2008) Modulation of C-reactive protein, tumor necrosis factor-alpha, and adiponectin by diet, exercise, and weight loss. J Nutr 138:2293–2296
Beasley LE, Koster A, Newman AB et al (2009) Inflammation and race and gender differences in computerized tomography-measured adipose depots. Obesity (Silver Spring) 17:1062–1069
Chandalia M, Cabo-Chan AV Jr, Devaraj S, Jialal I, Grundy SM, Abate N (2003) Elevated plasma high-sensitivity C-reactive protein concentrations in Asian Indians living in the United States. J Clin Endocrinol Metab 88:3773–3776
Muller S, Martin S, Koenig W et al (2002) Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-alpha or its receptors. Diabetologia 45:805–812
Burkart V, Kim YE, Hartmann B et al (2002) Cholera toxin B pretreatment of macrophages and monocytes diminishes their proinflammatory responsiveness to lipopolysaccharide. J Immunol 168:1730–1737
Lumeng CN, Deyoung SM, Saltiel AR (2007) Macrophages block insulin action in adipocytes by altering expression of signaling and glucose transport proteins. Am J Physiol Endocrinol Metab 292:E166–E174
Li L, Yang G, Shi S, Yang M, Liu H, Boden G (2009) The adipose triglyceride lipase, adiponectin and visfatin are downregulated by tumor necrosis factor-alpha (TNF-alpha) in vivo. Cytokine 45:12–19
Okada T, Liew CW, Hu J et al (2007) Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci USA 104:8977–8982
Palmer JP, Helqvist S, Spinas GA, Molvig J, Mandrup-Poulsen T, Andersen HU, Nerup J (1989) Interaction of beta-cell activity and IL-1 concentration and exposure time in isolated rat islets of Langerhans. Diabetes 38:1211–1216
Donath MY, Storling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T (2008) Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev 29:334–350
Kahn SE, Zraika S, Utzschneider KM, Hull RL (2009) The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia 52:1003–1012
Peterson JM, Pizza FX (2009) Cytokines derived from cultured skeletal muscle cells after mechanical strain promote neutrophil chemotaxis in vitro. J Appl Physiol 106:130–137
Yuen DY, Dwyer RM, Matthews VB et al (2009) Interleukin-6 attenuates insulin-mediated increases in endothelial cell signaling but augments skeletal muscle insulin action via differential effects on tumor necrosis factor-alpha expression. Diabetes 58:1086–1095
Mathur N, Pedersen BK (2008) Exercise as a mean to control low-grade systemic inflammation. Mediators Inflamm 2008:109502. doi:10.1155/2008/109502
Silva LA, Silveira PC, Pinho CA, Tuon T, Dal Pizzol F, Pinho RA (2008) N-acetylcysteine supplementation and oxidative damage and inflammatory response after eccentric exercise. Int J Sport Nutr Exerc Metab 18:379–388
Huey KA, McCusker RH, Kelley KW (2008) Exaggerated expression of skeletal muscle-derived interleukin-6, but not TNFalpha, in mice lacking interleukin-10. J Neuroimmunol 199:56–62
Meier-Ewert HK, Ridker PM, Rifai N et al (2004) Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol 43:678–683
Irwin MR, Wang M, Ribeiro D et al (2008) Sleep loss activates cellular inflammatory signaling. Biol Psychiatry 64:538–540
Maraldi C, Volpato S, Penninx BW et al (2007) Diabetes mellitus, glycemic control, and incident depressive symptoms among 70- to 79-year-old persons: the health, aging, and body composition study. Arch Intern Med 167:1137–1144
Pulkki-Raback L, Elovainio M, Kivimaki M et al (2009) Depressive symptoms and the metabolic syndrome in childhood and adulthood: a prospective cohort study. Health Psychol 28:108–116
Howren MB, Lamkin DM, Suls J (2009) Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 71:171–186
Raison CL, Borisov AS, Majer M et al (2009) Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry 65:296–303
McAfoose J, Baune BT (2009) Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev 33:355–366
Su S, Miller AH, Snieder H et al (2009) Common genetic contributions to depressive symptoms and inflammatory markers in middle-aged men: the Twins Heart Study. Psychosom Med 71:152–158
Tousoulis D, Drolias A, Antoniades C et al (2009) Antidepressive treatment as a modulator of inflammatory process in patients with heart failure: effects on proinflammatory cytokines and acute phase protein levels. Int J Cardiol 134:238–243
Larsen CM, Faulenbach M, Vaag A et al (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356:1517–1526
Berchtold LA, Larsen CM, Vaag A et al (2009) IL-1 receptor antagonism and muscle gene expression in patients with type 2 diabetes. Eur Cytokine Netw 20:81–87
Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T (2009) Sustained effects of interleukin-1-receptor antagonist treatment in type 2 diabetes mellitus. Diabetes Care 32:1663–1668
Aksentijevich I, Masters SL, Ferguson PJ et al (2009) An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 360:2426–2437
Channual J, Wu JJ, Dann FJ (2009) Effects of tumor necrosis factor-alpha blockade on metabolic syndrome components in psoriasis and psoriatic arthritis and additional lessons learned from rheumatoid arthritis. Dermatol Ther 22:61–73
Cheung D, Bryer-Ash M (2009) Persistent hypoglycemia in a patient with diabetes taking etanercept for the treatment of psoriasis. J Am Acad Dermatol 60:1032–1036
Manco M, Fernandez-Real JM, Equitani F et al (2007) Effect of massive weight loss on inflammatory adipocytokines and the innate immune system in morbidly obese women. J Clin Endocrinol Metab 92:483–490
Fleischman A, Shoelson SE, Bernier R, Goldfine AB (2008) Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31:289–294
Fernandez-Real JM, Lopez-Bermejo A, Ropero AB et al (2008) Salicylates increase insulin secretion in healthy obese subjects. J Clin Endocrinol Metab 93:2523–2530
Koska J, Ortega E, Bunt JC et al (2009) The effect of salsalate on insulin action and glucose tolerance in obese non-diabetic patients: results of a randomised double-blind placebo-controlled study. Diabetologia 52:385–393
Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, Shulman GI (2002) Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 109:1321–1326
Hotamisligil GS, Erbay E (2008) Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 8:923–934
Franceschi C, Capri M, Monti D et al (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 128:92–105
Dasu MR, Devaraj S, Zhao L, Hwang DH, Jialal I (2008) High glucose induces toll-like receptor expression in human monocytes: mechanism of activation. Diabetes 57:3090–3098
Schaeffler A, Gross P, Buettner R et al (2009) Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology 126:233–245
Chao W (2009) Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol 296:H1–H12
Ohashi K, Burkart V, Flohe S, Kolb H (2000) Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 164:558–561
Kim HJ, Higashimori T, Park SY et al (2004) Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 53:1060–1067
Acknowledgements
The authors regret that, because of space limitation, it was not possible to refer to all studies related to the topics of this review.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kolb, H., Mandrup-Poulsen, T. The global diabetes epidemic as a consequence of lifestyle-induced low-grade inflammation. Diabetologia 53, 10–20 (2010). https://doi.org/10.1007/s00125-009-1573-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1573-7