
Abstract
Aims/hypothesis
Inhibition of c-jun N-terminal kinase (JNK) favours pancreatic islet function and survival. Since two JNK isoforms are present in the pancreas (JNK1 and JNK2), we addressed their specific roles in experimental islet transplantation.
Methods
C57BL/6J (wild-type [WT]), Jnk1 (also known as Mapk8)−/− and Jnk2 (also known as Mapk9)−/− mice were used as donor/recipients in a syngeneic islet transplantation model. Islet cell composition, function, viability, production of cytokines and of vascular endothelial growth factor (VEGF) were also studied in vitro.
Results
Jnk1 −/− islets secreted more insulin in response to glucose and were more resistant to cytokine-induced cell death compared with WT and Jnk2 −/− islets (p < 0.01). Cytokines reduced VEGF production in WT and Jnk2 −/− but not Jnk1 −/− islets; VEGF blockade restored Jnk1 −/− islet susceptibility to cytokine-induced cell death. Transplantation of Jnk1 −/− or WT islets into WT recipients made diabetic had similar outcomes. However, Jnk1 −/− recipients of WT islets had shorter time to diabetes reversal (17 vs 55 days in WT, p = 0.033), while none of the Jnk2 −/− recipients had diabetes reversal (0% vs 71% in WT, p = 0.0003). Co-culture of WT islets with macrophages from each strain revealed a discordant cytokine production.
Conclusions/interpretation
We have shown a deleterious effect of JNK2 deficiency on islet graft outcome, most likely related to JNK1 activation, suggesting that specific JNK1 blockade may be superior to general JNK inhibition, particularly when administered to transplant recipients.
Similar content being viewed by others


Introduction
Islet cell transplantation is associated with the release of pro-inflammatory cytokines (CTKs), which play a key role in mediating the loss and functional impairment of beta cell mass via c-jun N-terminal kinase (JNK) activation, among other mechanisms [1–5]. Activation of JNK is usually reduced in culture, but immediately returns to high levels upon transplantation [6]. Although the pathway activated by CTKs and those activated by islet isolation may differ, JNK has been recognised as a key mediator involved in both [1]. Inhibition of JNK improves islet function and viability in experimental models of pancreatic islet transplantation [6–8]. A positive effect on recovery of islets [7, 8], glucose responsiveness [4, 8] and susceptibility to apoptosis at baseline and after exposure to CTKs [7, 9–11] has been reported. Thus, targeting JNK may improve functional beta cell mass via several mechanisms.
Three different isoforms of JNK (JNK1, 2 and 3) have been identified; JNK1 and JNK2 are widely produced, while JNK3 production is restricted to the brain [12]. In diabetes, JNK1 plays a very important role in obesity-induced insulin resistance [13], and JNK2 contributes to insulitis in NOD mice [14]. Furthermore, JNK1/2 co-inhibition prevents effector T cell function [15]. In Jnk2 (also known as Mapk9)−/− mice, a strong compensatory role of JNK1 has been described [16]. Thus, understanding the isoform-specific role of JNK in the context of islet isolation and transplantation may shed light on the mechanisms linking JNK to islet function and viability, and may allow for the development of novel therapeutic strategies for the preservation of beta cell mass and function after transplantation.
In the current study, we investigated the specific role of JNK1 and JNK2 isoforms in islet function and survival in vitro and in vivo. We have used Jnk1 (also known as Mapk8)−/− and Jnk2 −/− mice as donor or recipients of pancreatic islets in a syngeneic transplantation model. Although islets from Jnk1 −/− mice were characterised by a better in vitro function and survival profile when compared with wild-type (WT) islets, this effect did not translate into improved islet performance in vivo. When WT islets were transplanted into Jnk1 −/− or Jnk2 −/− mouse models of diabetes, a more rapid diabetes reversal was observed in Jnk1 −/− recipients, while none of the Jnk2 −/− recipient mice reverted diabetes. Overall, our data suggest that specific inhibition of JNK1 in islet transplant recipients may have beneficial effects on graft outcome.
Methods
Islet isolation and transplantation
Mapk8tm1Flv/J heterozygous (Jnk1 +/−) and Mapk9tm1Flv/J homozygous (Jnk2 −/−) mice on a C57BL6/J background were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Breeding and genotyping were performed as per vendor. C57BL6/J male mice served as syngeneic control (wild-type [WT]) of Jnk1 −/− and Jnk2 −/− mice. All animal procedures were conducted under protocols approved by the Institutional Animal Care and Use Committee and by the NIH. Streptozotocin (STZ) induction of diabetes and murine islet isolation and transplantation were performed as previously described [17]. The islet transplantation model consisted of a suboptimal islet mass (5,000 islet equivalents [IEQs]/kg) implanted under the kidney capsule of syngeneic diabetic recipients. Diabetes was induced by a single i.v. injection of STZ (200 mg/kg STZ; Sigma-Aldrich, St Louis, MO, USA). Diabetes occurrence was defined as non-fasting glycaemic values >19.5 mmol/l in three consecutive readings. WT, Jnk1 −/− and Jnk2 −/− mice were used as either donors or recipients of islets. Baseline fasting glycaemia and serum insulin level (Linco, Billerica, MA, USA) were determined. In the animals achieving normoglycaemia after transplantation, the graft-bearing kidney was removed under general anaesthesia in order to confirm that normoglycaemia was not a consequence of residual function of the native islets. For baseline intraperitoneal glucose tolerance tests (IPGTTs), an i.p. glucose bolus (2 g/kg) was administered after overnight fasting, and glycaemia was measured at baseline and over a 20 min period. Human islets from multi-organ deceased donors with research consent were used for selected in vitro experiments [18, 19].
CTK analysis
Islet-specific CTK production was tested both at baseline and after stimulation with a cocktail of exogenous CTKs for 18 h (IL-1β 50 U/ml, TNF-α 1,000 U/ml and INF-γ 1,000 U/ml; R&D Systems, Minneapolis, MN, USA). Briefly, cell supernatant fractions were collected, normalised for islet protein content, and CTK production analysed by Luminex technology (Bio-Rad, Hercules, CA, USA), these CTKs being IL-1β, INF-γ, TNF-α, monocyte chemoattractant protein 1 (MCP-1), RANTES (regulated upon activation normal T-expressed and secreted cytokine), macrophage inflammatory protein (MIP)-1α, MIP-1β, IL-6, IL-10 and vascular endothelial growth factor (VEGF). For experiments where islets received treatment with CTKs, data were expressed as percentage of untreated samples. Parallel experiments were performed in human islets treated after isolation with either TAT peptide alone or with the l-isoform of a TAT peptide inhibitor of JNK (L-JNKI, 10 μmol/l; BioSynthesis, Lewisville, TX, USA) and exposed for 18 h to CTKs as previously described [7].
Islet composition and cell viability assessment
Analysis of cellular composition was performed by immunofluorescence and quantified by laser scanning cytometry (LSC) as previously described [20]. Briefly, dispersed cells were incubated for 1 h with the following monoclonal antibodies: mouse to C-peptide (1:100; Abcam, Cambridge, MA, USA); rabbit to somatostatin (1:500; Sigma); mouse to glucagon (1:500; Sigma); and rabbit to pancreatic polypeptide (1:1,000; Sigma). After washing, samples were incubated with either goat anti-mouse or goat anti-rabbit (Alexa-Fluor-488 and Alexa-Fluor-647 IgG; Molecular Probes, Eugene, OR, USA). Omission of the primary antibody served as negative control. For the analysis of viability, CTKs were added 18 h prior to analysis. Briefly, the zinc-binding dye Newport Green (NG) allows quantification of islet beta cells on dissociated islets, based on the abundant zinc content in beta cell secretory granules [21]. The membrane exclusion dye 7-aminoactinomycin-D (7AAD; Invitrogen, Carlsbad, CA, USA) was used as a marker of cell death. Tetramethylrhodamine ethyl ester (TMRE; Molecular Probes) was used for the assessment of mitochondrial membrane potential. Single-cell suspensions were incubated with 110 μmol/l NG and 100 ng/ml TMRE for 30 min at 37°C and stained with 7AAD prior to analysis. NG+7AAD− cells that bound TMRE (TMRE+NG+7AAD−) were considered viable beta cells. Data are graphed as percentage of controls when comparing CTK-treated with untreated cells. For experiments requiring VEGF blockade, an anti-human VEGF neutralising antibody or an irrelevant isotype (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was added to the culture 18 h prior to viability assessment.
Islet cell function
Dynamic glucose-stimulated insulin release (GSIR) was tested as previously described [22]. Briefly, 100 IEQs per condition were loaded on micro-columns connected to an inflow and an outflow port of a customised perifusion system (BioRep Technologies, Miami, FL, USA). Media of defined composition (3 mmol/l glucose, 11 mmol/l glucose or 25 mmol/l KCl) were circulated at a rate of 100 μl/min, and samples collected every minute for the determination of insulin content by ELISA (Mercodia, Uppsala, Sweden).
Characterisation of total and phosphorylated JNK and c-jun in WT, Jnk1−/− and Jnk2−/− islets
Islets were collected in lysis buffer for the analysis of phosphorylated and total JNK and c-jun by western blot and Luminex technology (Bio-Plex), a fluorescence-based quantitative analysis of protein concentration. For the analysis of the abundance of the JNK1 and JNK2 isoforms, 35 μg of islet cell lysates was loaded onto each well of an SDS-PAGE gel (Bio-Rad), separated by electrophoresis and transferred onto a nitrocellulose membrane (Amersham Bioscience, Germany). The membrane was blocked in 5% (vol./vol.) non-fat milk (Carnation) in TRIS-buffered saline containing 0.1% (vol./vol.) Tween-20 for 1 h and then incubated with anti-JNK antibody (Cell Signaling, Danvers, MA, USA) at 4°C overnight followed by a horseradish peroxidase-conjugated goat anti-mouse (1:1,000 dilution; Pierce, Rockford, IL, USA) for 60 min at room temperature. Data obtained with Bio-Plex were validated in prior studies by western blotting and activity assay (Cell Signaling) [23].
Macrophages/islet co-culture and CTK production
Repeated peritoneal washes were performed in mice using RPMI-1640. Pooled mixtures were centrifuged and resuspended in NH4Cl (Stem-Cell Technologies, Vancouver, Canada) allowing for erythrocyte lysis. After two washes in RPMI with 5% (vol./vol.) FBS, 2 × 106 macrophages (MØs)/well were plated into 24 well plates. After 4 h incubation at 37°C, adherent MØs were stimulated with 20 mg/ml (100 U/ml) lipopolysaccharide (Sigma), and 100 U/ml recombinant murine IFN-γ (R&D Systems) and incubated for 18–72 h in the presence or absence of WT islets (100 IEQs/well). Supernatant fractions were collected at 48 h and assayed for CTKs as described above.
Data analysis
Results represent the mean of between four and eight independent experiments. Results are expressed as bar graphs with means and SDs. When one-way ANOVA showed statistical significance, results were compared using a t test after Tukey’s correction for multiple comparisons (GraphPad-Prism software). Statistical significance was set at p < 0.05. For in vivo experiments, between six and eight recipients per group were used, and all data from WT recipients of WT islets were pooled and used as control. Time to diabetes reversal was assessed using Kaplan–Meier survival curves and the log-rank test. Proportions were compared using χ 2 tests.
Results
Jnk1−/− islets have a better function than WT islets in vitro
We first analysed the in vitro function of WT islets treated with a L-JNKI as previously reported [23], and found enhanced GSIR in L-JNKI-treated islets when compared with islets treated with TAT peptide (Fig. 1a,c). In order to understand if such a beneficial effect was related to blockade of JNK1 or JNK2, we used Jnk1 −/− or Jnk2 −/− islets, and found a higher insulin secretion in Jnk1 −/− islets compared with WT (Fig. 1b,e). GSIR in Jnk2 −/− islets was not different from WT (Fig. 1c,f). In order to examine islet function in vivo, we performed IPGTTs in 18-week-old non-diabetic mice. Fasting glycaemia and serum insulin were identical among groups. In particular, fasting insulin levels were 28 ± 19 pmol/l in WT, 26.3 ± 3.3 in Jnk1 −/− and 30.6 ± 14 in Jnk2 −/− (p = NS). After glucose bolus, Jnk1 −/− and Jnk2 −/− mice showed similar glucose disposal profiles when compared with WT mice (AUC: 34.9 ± 2.1, 30.6 ± 6.8, 37.7 ± 6.3 mmol/l × min, respectively, p = NS). In order to understand if the different function of Jnk1 −/− islets in vitro was associated with a different content of endocrine cells, islet composition assessment was performed and was equal among groups (Table 1). When insulin was measured in lysates of freshly isolated islets, Jnk1 −/−, Jnk2 −/− and WT islets showed comparable insulin content (3.17 ± 1.23, 3.13 ± 0.22 and 3.26 ± 1.11 μmol/mg protein, respectively; p = NS).

Representative perifusion profile after exposure to 11 mmol/l glucose (G) and 25 mmol/l KCl are shown in the upper graphs (a–c). Below each perifusion, bar graphs of maximal values of insulin secretion in response to 11 mmol/l glucose from five independent experiments are shown (d–f). Data are expressed as pmol insulin/μg DNA and are presented as means and SDs. a, d WT islets treated with L-JNKI (dotted line) vs controls (solid line). b, e Jnk1 −/− islets (dotted line) vs WT (solid line). c, f Jnk2 −/− islets (dotted line) vs WT (solid line).*p < 0.05, **p < 0.01 vs WT
Jnk1−/− islets are resistant to CTK-induced cell death
Because beta cell viability is a major determinant of islet cell function and graft survival [20], and JNK is a major determinant of islet viability [2, 7, 8, 23], we investigated if Jnk1 −/− islets were more resistant to CTKs. We used a cocktail of CTKs that are important in leucocyte-mediated beta cell injury [24, 25] and that compromise islet cell viability at least partially via JNK [7, 11]. Viability at baseline was similar in all experimental groups. A ∾40% reduction in islet cell viability was observed after CTK exposure in both WT and Jnk2 −/− islets, while Jnk1 −/− islets appeared significantly protected (Fig. 2).

Viability of dissociated islet cells was evaluated as NG+7AAD− cells that bound TMRE (viable beta cells). WT, Jnk1 −/− and Jnk2 −/− islet cells were evaluated both at baseline (controls) and after CTK stimulation. Shown is a bar graph of the mean and SDs of four independent experiments. *p < 0.05, **p < 0.01
Jnk2−/− islets have higher ability to phosphorylate JNK and c-jun when compared with WT and Jnk1−/−
Since the ability of JNK1 to compensate for lack of JNK2 and vice versa has been described in peripheral tissues and lymphocytes, we tested if such compensation would occur in islets. We first determined the amount and the isoforms of JNK produced by either group (Fig. 3a). JNK1 was prevalent in WT islets. Lack of JNK1 resulted in a significant reduction of total JNK being produced by islets, while a compensatory increase in JNK1 was observed in Jnk2 −/− (Fig. 3a). Despite a similar degree of total JNK produced by Jnk2 −/− and WT islets, Jnk2 −/− islets were characterised by a higher degree of JNK (Fig. 3b) and c-jun (Fig. 3c) phosphorylation when compared with WT and Jnk1 −/− at baseline and after CTK stimulation (Fig. 3d). JNK1 was also the isoform most represented in kidney cortexes, and Jnk2 −/− kidney cortexes were also characterised by increased JNK phosphorylation (data not shown).

a Representative blot analysis of JNK1 (45 kDa) and JNK2 (54 kDa) isoform production in islets isolated from Jnk1 −/−, WT and Jnk2 −/− mice, and quantitative evaluation by fluorescence intensity of total JNK per mg of protein in WT, Jnk1 −/− and Jnk2 −/− islets. b Evaluation of fluorescence of phosphorylated (phospho)/total JNK in WT, Jnk1 −/− and Jnk2 −/− islets. c Evaluation of fluorescence of phospho/total c-jun in WT, Jnk1 −/− and Jnk2 −/− islets. d Evaluation of fluorescence of phospho/total JNK after exposure to CTK in WT, Jnk1 −/− and Jnk2 −/− islets. Data are the means and SDs of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
Jnk1−/− and Jnk2−/− islets produce less MCP-1 than WT islets
In order to analyse if the resistance to cell death was related to a different CTK production, islet-specific CTK production was evaluated in the supernatant fractions of WT, Jnk1 −/− and Jnk2 −/− islets. INF-γ and IL-10 were not detectable, with a limit of detectability that was 3 pg/ml for IL-10 and 1.9 pg/ml for INF-γ. Jnk1 −/− and Jnk2 −/− islets were both characterised by a significantly lower MCP-1 production than WT islets (2.21 and 1.61 vs 4.23 nmol/l, respectively, p < 0.05). All other CTKs (IL-1β, TNF-α, RANTES, MIP-1α, MIP-1β, IL-6 and VEGF) were detectable but not different amongst groups (data not shown). Upon CTK stimulation, both Jnk1 −/− and Jnk2 −/− islets were significantly protected from an increase in MCP-1 production (Fig. 4a). Treatment with L-JNKI in parallel experiments in human islets reduced MCP-1 production both at baseline and after CTK stimulation (Fig. 4b).

Bar graphs of MCP-1 (a) and VEGF (c) secretion in WT, Jnk1 −/− and Jnk2 −/− islets exposed to CTK compared with secretion in untreated islets. The data are the means and SDs of five independent experiments. The effects of a general JNK inhibitor (L-JNKI) administered to cultured human islets on MCP-1 (b) and VEGF (d) production are also shown. L-JNKI prevented MCP-1 production and facilitated VEGF secretion both in controls islets (C) and after CTK exposure. *p < 0.05, **p < 0.01
Increased VEGF production by Jnk1−/− islets is associated with their higher viability
VEGF production at baseline was equal amongst experimental groups (86.5 ± 24.5, 70.2 ± 14.6, 80.01 ± 8.4 nmol/l in WT, Jnk1 −/− and Jnk2 −/−, respectively). However, CTK treatment resulted in decreased VEGF production in both WT and Jnk2 −/− islets, but not in Jnk1 −/− islets (Fig. 4c). When a general JNK inhibitor was applied to human islets, however, L-JNKI resulted in an improvement of VEGF release both at baseline and after CTK exposure (Fig. 4d). Given the preserved viability and VEGF production in CTK-treated Jnk1 −/− islets, we repeated viability experiments in Jnk1 −/− islets in the presence of a VEGF-neutralising antibody, and we found that abrogation of VEGF action in Jnk1 −/− islets restores their susceptibility to CTK-induced cell death (Fig. 5). Administration of a VEGF-neutralising antibody to CTK-treated WT islets did not further compromise cell death (data not shown).

Viability of Jnk1 −/− islet cells was evaluated as NG+7AAD− cells that bound TMRE (viable beta cells) in the absence (−) or presence (+) of CTK alone or combined with a VEGF-neutralising antibody (VEGF-Ab). Data are the means and SDs of four independent experiments and are expressed as a percentage of the viability of islets exposed to an irrelevant antibody control. **p < 0.01
Jnk1−/− islets do not retain a survival advantage after transplantation
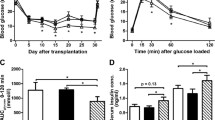
When islets from either WT, Jnk1 −/− or Jnk2 −/− mice were transplanted into WT diabetic mice, no significant differences in graft performance (namely, percentage of mice achieving euglycaemia over time) was observed amongst groups (Fig. 6a,b). All animals reverted to diabetes after surgical nephrectomy of the graft-bearing kidney.

The graft outcomes of syngeneic marginal mass transplantation experiments. The percentage of diabetic mice was plotted against time after transplantation for the following set of experiments: a Jnk1 −/− (dotted line) or WT islets (solid line) into WT recipients; b Jnk2 −/− (dotted line) or WT islets (solid line) into WT recipients; c WT islets into Jnk1 −/− (dotted line) or WT recipients (solid line); d WT islets into Jnk2 −/− (dotted line) or WT recipients (solid line)
JNK1 deficiency in diabetic recipients of WT islets is beneficial for graft performance, while JNK2 deficiency is deleterious
When WT islets were transplanted into diabetic WT, Jnk1 −/− or Jnk2 −/− recipients, median time to diabetes reversal was different between WT and Jnk1 −/− mice (17 vs 55 days, p = 0.033, Fig. 6c). On the contrary, the proportion of Jnk2 −/− mice with diabetes reversal after transplantation was significantly lower than in WT mice (0% vs 71%, p = 0.0003, Fig. 6d). All animals become equally hyperglycaemic after nephrectomy of the graft-bearing kidney. Haematoxylin and eosin staining of the marginal mass graft showed a preserved islet anatomy in WT and Jnk1 −/− recipients (Fig. 7a,b), while the architecture of islets transplanted into Jnk2 −/− recipients was totally disrupted (Fig. 7c) and insulin staining was undetectable (data not shown).

Representative ×20 magnifications of haematoxylin and eosin staining of islets graft are shown for WT islets into WT recipients (a), WT islets into Jnk1 −/− mice (b) and WT islets into Jnk2 −/− mice (c). To normalise for the effect of glycaemia on graft morphology, we have elected to show the graft histology from mice that were equally hyperglycaemic
CTK production by WT islets co-cultured with WT, Jnk1−/− or Jnk2−/−MØs
In order to investigate one potential mechanism responsible for the opposite graft survival observed between Jnk1 −/− and Jnk2 −/− recipients of WT islets, we performed in vitro co-culture experiments between WT islets and WT, Jnk1 −/− or Jnk2 −/− MØs. We first compared CTK production by WT, Jnk1 −/− or Jnk2 −/− MØs at baseline and after activation (lipopolysaccharide+INF-γ). A lower production of IL-1β was observed in Jnk1 −/− activated MØs when compared with WT or Jnk2 −/− MØs (360 ± 119 vs 773.3 ± 173.3 vs 712.7 ± 80.6 nmol/l, respectively, p < 0.01). Co-culture of WT islets with Jnk1 −/− or Jnk2 −/− activated MØs resulted in less IL-1β, MCP-1 and IL-6 production when compared with WT islets co-cultured with WT MØs (Fig. 8). Interestingly, TNF-α production was discordant when Jnk1 −/− and Jnk2 −/− MØs where co-cultured with WT islets.

CTK production by WT islets co-cultured with WT, Jnk1 −/− or Jnk2 −/− MØs. While IL-1β (a), MCP-1 (b) and IL-6 (c) production were markedly reduced when either Jnk1 −/− and Jnk2 −/− MØs were co-cultured with WT islets and compared with WT MØs co-cultured with WT islets, an opposite effect on TNF-α production (d) was observed when Jnk1 −/− or Jnk2 −/− MØs were used, with higher levels of TNF-α produced when Jnk2 −/− MØs were used. The bar graphs show the mean and SDs of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Several beneficial effects of JNK inhibitors on pancreatic islets have been reported [6, 7]. Although a redundant function of JNK1 and JNK2 has been described [12], certain functions, such as TNF-α-induced apoptosis, operate via JNK1 only [26]. In the present study, we show that JNK1 gene knock-out is more beneficial than JNK2 gene knock-out on islet cell function and survival in vitro. Furthermore, Jnk1 −/− mouse recipients of WT islets had faster diabetes reversal than WT, while all Jnk2 −/− recipients remained diabetic and were characterised by a strong JNK activation through compensatory mechanisms. Our data suggest an important role of JNK1 on pancreatic islet graft outcome. The major strength of our findings is that we provide a first analysis of the role of different JNK isoforms in islets function and viability. Major weaknesses are the inability to identify a specific mechanism responsible for the in vivo loss of in vitro benefits, the use of only syngeneic models of transplantation, and the limited therapeutic usefulness of our findings due to high sequence and structure similarities between different isoforms [12, 27]. An additional limitation is the use of JNK isoform knock-out animal models, where known compensatory effects occur [16, 28].
JNK inhibition ameliorates islet cell function in vitro [2, 29]. We found that treatment with a general JNK inhibitor as well as JNK1 deficiency led to a significant increase of insulin release in the first phase of insulin secretion (Fig. 1a,b) without affecting cellular composition (Table 1). Interestingly, when evaluating glucose responsiveness in vivo during IPGTTs, no significant difference in glucose clearance was observed between Jnk1 −/−, Jnk2 −/− and WT mice. Thus, the in vitro functional benefit of Jnk1 −/− islets was not reflected by a better glucose responsiveness in vivo, suggesting that systemic pathways regulated by JNK may overcome the local islet-specific effects.
Islet graft function after transplantation is strongly affected by the exposure to inflammatory mediators at the implantation site. Thus, we proceeded to investigate if Jnk1 −/− or Jnk2 −/− islets were more resistant to the effect of CTK on cell viability when compared with WT islets. We found a complete resistance of Jnk1 −/− islets to the effect of CTKs, while both WT and Jnk2 −/− islets viability was reduced to 60% of that of control untreated cells (Fig. 2). This result is in contrast to a recent report suggesting that cell death is comparable between WT and Jnk1 −/− islets exposed to CTKs, where both samples were associated with a sevenfold increase in the rate of cell death [10]. It is conceivable that a milder noxious stimulus than the one we have used (IL-1β at 14.7 instead of 588.2 pmol/l, 18 h instead of 24 h) may be necessary to observe the protective role of Jnk1 −/− deficiency. Although our data may initially suggest an opposite function of JNK1 and JNK2 in islets cells, the evidence that Jnk2 −/− islets are characterised by a strong overcompensation by JNK1 may have masked the beneficial effect of lack of JNK2 and be responsible for the similar behaviours observed in vitro in WT and Jnk2 −/− islets (Fig. 3). Since CTKs released by islets may also negatively affect graft outcome [30, 31], we investigated the CTK concentration in culture supernatant fractions from WT, Jnk1 −/− and Jnk2 −/− islets. Both Jnk1 −/− and Jnk2 −/− islets produced less MCP-1 than WT, and MCP-1 blockade facilitates survival of islet allografts [32]. The levels of MCP-1 production remained lower in Jnk1 −/− and Jnk2 −/− than in WT islets even after exposure to CTKs (Fig. 4a), similar to what we observed in human islets treated with L-JNKI (Fig. 4b). Our findings are consistent with prior reports on rat vascular smooth muscle cells, where JNK inhibition prevented TNF-α-induced upregulation of MCP-1 [33]. The fact that both Jnk1 −/− and Jnk2 −/− islets prevented CTK-mediated MCP-1 upregulation, while only Jnk1 −/− islets were resistant to the negative effect of exogenous CTK on viability, suggests that MCP-1 per se did not contribute to islet susceptibility to cell death in our model. Among other CTKs, VEGF production by islet cells was compromised in WT and Jnk2 −/− but not in Jnk1 −/− islets (Fig. 4c). This was an interesting finding, because VEGF has been associated with increased functional beta cell mass [34]. Since VEGF has been directly implicated in cell survival [35, 36], we investigated if VEGF blockade could reverse the resistance of Jnk1 −/− islets to CTK-induced cell death. We were able to demonstrate that treatment of Jnk1 −/− islets with a VEGF-neutralising antibody was able to restore Jnk1 −/− susceptibility to cell death (Fig. 5). To the best of our knowledge, this is the first evidence that VEGF may have a direct effect on islet cell viability.
The observed functional and survival benefit of Jnk1 −/− islets over WT or Jnk2 −/− islets would suggest a potential advantages for in vivo application in the transplantation setting. To our surprise, we did not detect any difference in the performance of syngeneic marginal islet mass grafts when WT, Jnk1 −/− or Jnk2 −/− islets were transplanted into diabetic WT recipients (Fig. 6a,b). Thus, the early events in the graft microenvironment probably overcome the observed in vitro benefit of islet-specific JNK deficiency. In agreement with this hypothesis is our previous observation that L-JNKI can significantly protect human islets in vitro but that improved performance in vivo after transplantation is measurable only when diabetic recipients are treated as well [7]. In addition, recent data using an intrahepatic rodent model of syngeneic islet transplantation point to the important role for JNK inhibition in the immediate post-transplantation period [6]. In order to investigate the specific role of JNK1 and JNK2 in the modulation of the local inflammatory response at the site of islet transplantation, we performed experiments where WT islets obtained from the same isolation procedure were transplanted into diabetic WT, Jnk1 −/− or Jnk2 −/− recipients. Our data indicate that lack of JNK1 in the recipient microenvironment results in improved graft performance (measured as a faster time to diabetes reversal, Fig. 6c). Inversely, lack of JNK2 in the recipient’s microenvironment resulted in a complete inability to revert diabetes (Fig. 6d). Analysis of the graft at killing, i.e. 90 days after transplantation, failed to reveal a highly significant inflammatory infiltrate, although the islets architecture was totally disrupted when transplanted into Jnk2 −/− recipients and insulin was not any longer detectable (Fig. 7). It is possible that the activation of JNK observed in the kidney cortex of Jnk2 −/− mice (similar to what we showed for islets in Fig. 3) is responsible for the worse outcome, suggesting the importance of local environmental factors. Alternatively JNK2 is an important mediator of autoimmune-mediated islet injury [14], but may have a different role in the modulation of early local inflammatory responses occurring in syngeneic transplantation. Finally, it is possible that a similar compensatory mechanism occurred in Jnk2 −/− MØs, which could explain way the production of TNF-α was higher in Jnk2 −/− MØs and lower in Jnk1 −/− MØs when compared with WT (Fig. 8), although it has been suggested that Jnk2 −/− MØs are unable to phosphorylate c-jun [37]. Our data are consistent with a recent report suggesting that JNK1 signalling may play an essential role in MØ-induced beta cell death when a multiple low-dose STZ model of pancreatic injury is used [10]. In this report, Jnk1 −/− mice were characterised by a strikingly reduced production of TNF-α by MØs. Although JNK2 is an important mediator of autoimmune-mediated islet injury [14], it may have a different role in the modulation of early local inflammatory responses occurring in syngeneic transplantation. In order to investigate the specific role of recipient MØs on graft function in our model, we elected to study how the co-culture of WT islets with activated WT, Jnk1 −/− or Jnk2 −/− MØs would affect CTK release. A reduced release of IL-1β, MCP-1 and IL-6 was observed in Jnk1 −/− or Jnk2 −/− MØs when compared with WT MØs (Fig. 8). Interestingly, while WT islets incubated with Jnk1 −/− MØs resulted in reduced TNF-α production, the opposite was observed for Jnk2 −/− MØs. Our observations, together with a recent report in human prostate cancer cells showing that JNK1 but not JNK2 is the mediator of TNF-induced reactive oxygen species generation [38], strongly support a specific role of JNK1 in the regulation of TNF-α production. Future studies are warranted to investigate the specific role of JNK1 and JNK2 in allogeneic models of islet transplantation.
In summary, we have provided evidence that lack of JNK1 in murine islets results in improved function and cell viability. The improved viability of Jnk1 −/− islet in vitro was at least partially explained by a preserved VEGF production after CTK exposure. Such in vitro advantage was no longer evident in syngeneic transplantation models. However, Jnk1 −/− recipients of WT islets displayed an improved graft performance when compared with WT. On the contrary, none of the Jnk2 −/− recipients of WT islets reverted diabetes, probably through a compensatory overactivation of JNK1. Collectively, our data suggest that JNK1 and JNK2 may affect islet cell grafts in a very different manner, and that selective targeting of JNK1 in islet transplant recipients may represent a viable strategy to improve graft outcome.
Abbreviations
- 7AAD:
-
7-aminoactinomycin-D
- CTK:
-
cytokine
- GSIR:
-
glucose-stimulated insulin release
- IEQ:
-
islet equivalent
- IPGTT:
-
intraperitoneal glucose tolerance test
- JNK:
-
c-jun N-terminal kinase
- L-JNKI:
-
cell-permeable TAT peptide inhibitor of JNK
- LSC:
-
laser scanning cytometry
- MØ:
-
macrophage
- MCP-1:
-
monocyte chemoattractant protein 1
- MIP:
-
macrophage inflammatory protein
- NG:
-
Newport Green
- RANTES:
-
regulated upon activation normal T-expressed and secreted cytokine
- STZ:
-
streptozotocin
- TMRE:
-
tetramethylrhodamine ethyl ester
- VEGF:
-
vascular endothelial growth factor
- WT:
-
wild-type
References
Abdelli S, Ansite J, Roduit R et al (2004) Intracellular stress signaling pathways activated during human islet preparation and following acute cytokine exposure. Diabetes 53:2815–2823
Ammendrup A, Maillard A, Nielsen K et al (2000) The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 49:1468–1476
Rosenberg L, Wang R, Paraskevas S, Maysinger D (1999) Structural and functional changes resulting from islet isolation lead to islet cell death. Surgery 126:393–398
Aikin R, Maysinger D, Rosenberg L (2004) Cross-talk between phosphatidylinositol 3-kinase/AKT and c-jun NH2-terminal kinase mediates survival of isolated human islets. Endocrinology 145:4522–4531
Paraskevas S, Aikin R, Maysinger D et al (1999) Activation and expression of ERK, JNK, and p38 MAP-kinases in isolated islets of Langerhans: implications for cultured islet survival. FEBS Lett 455:203–208
Noguchi H, Nakai Y, Ueda M et al (2007) Activation of c-Jun NH(2)-terminal kinase (JNK) pathway during islet transplantation and prevention of islet graft loss by intraportal injection of JNK inhibitor. Diabetologia 50:612–619
Fornoni A, Pileggi A, Molano RD et al (2008) Inhibition of c-jun N terminal kinase (JNK) improves functional beta cell mass in human islets and leads to AKT and glycogen synthase kinase-3 (GSK-3) phosphorylation. Diabetologia 51:298–308
Noguchi H, Nakai Y, Matsumoto S et al (2005) Cell permeable peptide of JNK inhibitor prevents islet apoptosis immediately after isolation and improves islet graft function. Am J Transplant 5:1848–1855
Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77–82
Fukuda K, Tesch GH, Nikolic-Paterson DJ (2008) c-Jun amino terminal kinase 1 deficient mice are protected from streptozotocin-induced islet injury. Biochem Biophys Res Commun 366:710–716
Abdelli S, Abderrahmani A, Hering BJ, Beckmann JS, Bonny C (2007) The c-Jun N-terminal kinase JNK participates in cytokine- and isolation stress-induced rat pancreatic islet apoptosis. Diabetologia 50:1660–1669
Bogoyevitch MA (2006) The isoform-specific functions of the c-Jun N-terminal kinases (JNKs): differences revealed by gene targeting. Bioessays 28:923–934
Hirosumi J, Tuncman G, Chang L et al (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Jaeschke A, Rincon M, Doran B et al (2005) Disruption of the Jnk2 (Mapk9) gene reduces destructive insulitis and diabetes in a mouse model of type I diabetes. Proc Natl Acad Sci USA 102:6931–6935
Dong C, Yang DD, Tournier C et al (2000) JNK is required for effector T-cell function but not for T-cell activation. Nature 405:91–94
Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS (2006) Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci USA 103:10741–10746
Pileggi A, Molano RD, Berney T et al (2001) Heme oxygenase-1 induction in islet cells results in protection from apoptosis and improved in vivo function after transplantation. Diabetes 50:1983–1991
Ricordi C, Strom TB (2004) Clinical islet transplantation: advances and immunological challenges. Nat Rev Immunol 4:259–268
Ichii H, Pileggi A, Molano RD et al (2005) Rescue purification maximizes the use of human islet preparations for transplantation. Am J Transplant 5:21–30
Ichii H, Inverardi L, Pileggi A et al (2005) A novel method for the assessment of cellular composition and beta-cell viability in human islet preparations. Am J Transplant 5:1635–1645
Lukowiak B, Vandewalle B, Riachy R et al (2001) Identification and purification of functional human beta-cells by a new specific zinc-fluorescent probe. J Histochem Cytochem 49:519–528
Brendel MD, Kong SS, Alejandro R, Mintz DH (1994) Improved functional survival of human islets of Langerhans in three-dimensional matrix culture. Cell Transplant 3:427–435
Fornoni A, Cobianchi L, Sanabria NY et al (2007) The l-isoform but not d-isoforms of a JNK inhibitory peptide protects pancreatic beta-cells. Biochem Biophys Res Commun 354:227–233
Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, Svenson M (1986) Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science 232:1545–1547
Mandrup-Poulsen T, Bendtzen K, Dinarello CA, Nerup J (1987) Human tumor necrosis factor potentiates human interleukin 1-mediated rat pancreatic beta-cell cytotoxicity. J Immunol 139:4077–4082
Liu J, Minemoto Y, Lin A (2004) c-Jun N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for tumor necrosis factor alpha-induced c-Jun kinase activation and apoptosis. Mol Cell Biol 24:10844–10856
Bogoyevitch MA, Arthur PG (2008) Inhibitors of c-Jun N-terminal kinases: JuNK no more. Biochim Biophys Acta 1784:76–93
Jaeschke A, Karasarides M, Ventura JJ et al (2006) JNK2 is a positive regulator of the c-Jun transcription factor. Mol Cell 23:899–911
Lee YH, Giraud J, Davis RJ, White MF (2003) c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 278:2896–2902
Mandrup-Poulsen T (2003) Beta cell death and protection. Ann N Y Acad Sci 1005:32–42
Amoli MM, Larijani B (2006) Would blockage of cytokines improve the outcome of pancreatic islet transplantation. Med Hypotheses 66:816–819
Lee I, Wang L, Wells AD et al (2003) Blocking the monocyte chemoattractant protein-1/CCR2 chemokine pathway induces permanent survival of islet allografts through a programmed death-1 ligand-1-dependent mechanism. J Immunol 171:6929–6935
Chen YM, Chiang WC, Lin SL, Wu KD, Tsai TJ, Hsieh BS (2004) Dual regulation of tumor necrosis factor-alpha-induced CCL2/monocyte chemoattractant protein-1 expression in vascular smooth muscle cells by nuclear factor-kappaB and activator protein-1: modulation by type III phosphodiesterase inhibition. J Pharmacol Exp Ther 309:978–986
Lai Y, Schneider D, Kidszun A et al (2005) Vascular endothelial growth factor increases functional beta-cell mass by improvement of angiogenesis of isolated human and murine pancreatic islets. Transplantation 79:1530–1536
Calvani M, Trisciuoglio D, Bergamaschi C, Shoemaker RH, Melillo G (2008) Differential involvement of vascular endothelial growth factor in the survival of hypoxic colon cancer cells. Cancer Res 68:285–291
Xie X, Cao F, Sheikh AY et al (2007) Genetic modification of embryonic stem cells with VEGF enhances cell survival and improves cardiac function. Cloning Stem Cells 9:549–563
Lu Z, Serghides L, Patel SN et al (2006) Disruption of JNK2 decreases the cytokine response to Plasmodium falciparum glycosylphosphatidylinositol in vitro and confers protection in a cerebral malaria model. J Immunol 177:6344–6352
Antosiewicz J, Ziolkowski W, Kaczor JJ, Herman-Antosiewicz A (2007) Tumor necrosis factor-alpha-induced reactive oxygen species formation is mediated by JNK1-dependent ferritin degradation and elevation of labile iron pool. Free Radic Biol Med 43:265–270
Acknowledgements
We acknowledge valuable assistance from the Juvenile Diabetes Research Foundation (JDRF)-supported Preclinical Cell Processing and Imaging Cores (JDRF grant 4-2004-361). We also acknowledge the superb assistance of the Diabetes Research Institute (DRI) cGMP Human Cell Processing Facility and the National Center for Research Resources (NCRR)-sponsored Islet Cell Resources that provided access to human islets. The current study was supported by a pilot project (A. Fornoni) of a JDRF Center Grant 4-2004-361 (C. Ricordi), and by the DRI Foundation. The authors are grateful to E. Zahr, J. Molina, S. San Jose, Y. Gadea and K. Johnson from the DRI for outstanding technical assistance.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Varona-Santos, J.L., Pileggi, A., Molano, R.D. et al. c-Jun N-terminal kinase 1 is deleterious to the function and survival of murine pancreatic islets. Diabetologia 51, 2271–2280 (2008). https://doi.org/10.1007/s00125-008-1169-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1169-7