
Abstract
Aims/hypothesis
Orexin/hypocretin is a hypothalamic neuropeptide that regulates motivated behaviours, such as feeding and arousal, and, importantly, is also involved in energy homeostasis. The aim of this study was to reveal the role of orexin in the regulation of insulin sensitivity for glucose metabolism.
Methods
Orexin knockout mice fasted overnight underwent oral glucose tolerance testing and insulin tolerance testing. The impact of orexin deficiency on insulin signalling was studied by Western blotting to measure levels of Akt phosphorylation and its upstream and downstream molecules in the hypothalamus, muscle and liver in orexin knockout mice.
Results
We found that orexin deficiency caused the age-related development of impaired glucose tolerance and insulin resistance in both male mice without obesity and female mice with mild obesity, fed a normal chow diet. When maintained on a high-fat diet, these abnormalities became more pronounced exclusively in female orexin knockout mice that developed severe obesity. Insulin signalling through Akt was disrupted in peripheral tissues of middle-aged (9-month-old) but not young adult (2-to-3-month-old) orexin knockout mice fed a normal chow diet. Moreover, basal levels of hypothalamic Akt phosphorylation were abnormally elevated in orexin knockout mice at every age studied, and insulin stimulation failed to increase the level of phosphorylation. Similar abnormalities were observed with respect to GSK3β phosphorylation in the hypothalamus and peripheral tissues of middle-aged orexin knockout mice.
Conclusions/interpretation
Our results demonstrate a novel role for orexin in hypothalamic insulin signalling, which is likely to be responsible for preventing the development of peripheral insulin resistance with age.
Similar content being viewed by others


Introduction
The hypothalamus contains multiple neuronal systems responsible for the regulation of energy homeostasis. Among them, considerable attention has been focused on orexin, also known as hypocretin, because of its potent regulation of wakefulness, feeding behaviour and energy metabolism [1]. Orexin-A and -B are a pair of neuropeptides cleaved from a common precursor peptide, prepro-orexin [2, 3]. Orexin neurons are localised in the posterolateral hypothalamus and project into the entire brain except the cerebellum [1]. Electrophysiological studies on hypothalamic slices have shown that orexin neurons are responsive to peripheral metabolic cues [4, 5]. The excitability of orexin neurons was inhibited by glucose and leptin, indicators of high energy levels, whereas ghrelin, a signal for energy depletion, caused a stimulatory effect. The glucosensing machinery of orexin neurons is sufficiently sensitive to translate physiological glucose variations into different firing rates through tandem-pore K+ channels [6]. In fact, in vivo, orexin neurons effectively respond to nutritional status [7], and levels of mRNA transcripts for prepro-orexin are increased upon fasting or insulin-induced hypoglycaemia in rats [3, 8]. Thus, hypothalamic orexin neurons are a central site for integrating peripheral metabolic signals.
An association between short habitual sleep duration and increased BMI has been found in a large cohort study, suggesting interaction between the homeostatic systems regulating sleep/wakefulness and energy balance [9]. Orexin deficiency has been shown to be involved in the pathogenesis of narcolepsy in human and animal models, such as mice with knockout mutations in the gene for prepro-orexin (prepro-orexin knockout mice, referred to throughout the remainder of this article as orexin knockout mice) and orexin neuron-deficient mice [10, 11]. In humans, narcolepsy is accompanied by energy imbalance, such as decreased energy intake and increased BMI, resulting in an increase in the incidence of type 2 diabetes [12–15]. Mice deficient in orexin neurons also exhibit late-onset obesity, despite eating less [16], and on a high-fat diet, become heavier than wild-type mice [17]. Thus, orexin neurons appear to provide a crucial link between arousal and energy balance [1, 11, 18].
Circulating levels of glucose, a major source of energy, are tightly regulated by a sophisticated mechanism in which insulin plays a critical role by reducing hepatic glucose production and stimulating glucose transport into muscle and fat [19, 20]. Insulin is also present in mammalian brain [21], and hypothalamic insulin actions cause inhibition of hepatic glucose production, which regulates fasting blood glucose levels [22, 23]. We have previously reported that central administration of orexin reduces fasting blood glucose levels in mice, without changing serum insulin levels [24]. It is therefore possible that orexin enhances the actions of insulin in the hypothalamus to reduce fasting glucose, although the interaction between orexin and insulin in this region of the brain has not been clarified. In this study, we investigated the effect of orexin deficiency on glucose/insulin homeostasis using orexin knockout mice, and provide the first evidence that orexin is an essential factor for maintaining hypothalamic and peripheral insulin sensitivity for glucose metabolism, particularly with advancing age.
Methods
Animals
Orexin knockout mice with a mixed C57BL/6J–129/SvEv background, which completely lack both orexin-A and -B, were generated using a standard method, as described previously [25]. N5 or N10 generations of heterozygous (+/−) mice (backcrossed to C57BL/6J mice) were mated to obtain null (−/−) mutants and wild-type (+/+) littermates. The N5 mice were used in this study unless otherwise indicated. The offspring were genotyped by PCR using the primer set shown in Electronic supplementary material (ESM) Table 1. The animals were housed under standard light (12 h light/dark cycle) and temperature conditions, and were given free access to a normal diet (CE-2; Clea Japan, Tokyo, Japan) and water. Certain groups of mice were given a high-fat diet containing 30% fat (Rodent Quick Fat; Clea Japan) for 6 months or containing 60% fat (D12492; Research Diets, New Brunswick, NJ, USA) for 6 weeks. All experimental procedures were approved by the University of Toyama Animal Research Committee.
OGTT and insulin tolerance test
For the OGTT, mice fasted for 10 h were orally loaded with glucose (2 g/kg). For the insulin tolerance test, mice fasted for 10 h were intraperitoneally injected with human regular insulin (Humalin R; Eli Lilly, Indianapolis, IN, USA; 1 U/kg). Blood was collected from the tail vein, and blood glucose concentrations were determined using a glucose analyser (Antsense II, Horiba Industry, Kyoto, Japan).
Measurement of locomotor activity
Mice were placed individually in an acrylic cage, and after 1 h habituation, the locomotor activity (number of movements) was recorded at 30 min intervals for 24 h using a Scanet MV-20plus system (Melquest, Toyama, Japan).
Measurement of serum parameters
Blood was collected from the orbital sinus of fasted mice under halothane anaesthesia, and then kept on ice for 2 h before being centrifuged at 16,000 ×g for 1 min at 4°C. Serum was stored at −80°C until use. The serum insulin and leptin concentrations were analysed using an ELISA kit (Morinaga Seikagaku, Tokyo, Japan).
Intracerebroventricular administration of insulin
Mice were anaesthetised with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.) and positioned in a stereotaxic frame. A 10 mm long guide cannula was implanted into the lateral ventricle (0.3 mm posterior to bregma, 0.9 mm lateral from the central suture) and fixed to the skull with dental cement. The injection depth was 2.4 mm from the skull surface. After a 7 day recovery period, the mice were intracerebroventricularly injected with 3.0 μl of human regular insulin (10 mU per mouse).
Western blotting
Mice fasted overnight were given an i.v. tail vein injection (4 U/kg) or intracerebroventricular injection (10 mU) of human regular insulin, and killed 15 or 5 min after the injection, respectively. PBS was injected as vehicle control. Subsequently, the hypothalamus, hindlimb muscle and liver were dissected, frozen in liquid nitrogen, and stored at −80°C until use. The tissues were lysed and subjected to immunoprecipitation and/or Western blotting, as described previously [26]. Primary antibodies against Akt1, insulin receptor β (Santa Cruz Biotechnology, Santa Cruz, CA, USA); phosphotyrosine (PY99; Santa Cruz Biotechnology); glycogen synthase kinase 3β (GSK3β) (BD Transduction Laboratories, San Jose, CA, USA); IRS-1 and IRS-2 (Upstate Biotechnology, Lake Placid, NY, USA); AMP kinase α, phospho-Akt (Ser473), phospho-Akt (Thr308), phospho-AMP kinase α (Thr172), phospho-GSK3β (Ser9), p44/p42 mitogen-activated protein (MAP) kinase and phospho-p44/p42 MAP kinase (Thr202/Tyr204) (Cell Signaling Technology, Beverly, MA, USA); GLUT4 (Abcam, Cambridge, MA, USA); and β-actin (Sigma, St Louis, MO, USA) were used.
Statistical analysis
Data are represented as means±SEM. The significance of differences between two groups was assessed by Student’s t test, and differences between multiple groups were assessed by one-way ANOVA and Bonferroni test, using Statview software (SAS Institute, Cary, NC, USA). A p value of less than 0.05 was considered significant.
Results
Impaired glucose tolerance and insulin resistance in orexin deficiency
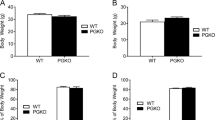
When maintained on a normal chow diet, male orexin knockout mice showed a similar growth pattern to their wild-type littermates from 4–36 weeks of age (Fig. 1a). Female orexin knockout mice, however, became 1.2-fold heavier than wild-type female mice at 36 weeks of age (Fig. 1b). Moreover, female, but not male, orexin knockout mice exhibited an increase in epididymal fat pad weight at 12 months of age compared with sex- and age-matched wild-type mice given unrestricted access to standard chow (Fig. 1a,b insets). The weights of soleus muscle, liver and pancreas were not different between wild-type and orexin knockout female mice at 9–12 months of age (ESM Table 2). These results indicate that female, but not male, orexin knockout mice become obese at middle age. Food intake was significantly reduced in 9- to 12-month-old orexin knockout female mice compared with wild-type female mice (ESM Table 2), indicating that obesity in female orexin knockout mice is not due to excessive food intake. Spontaneous locomotor activity during dark phase, but not light phase, was greatly reduced in male (data not shown) and female (ESM Table 2) orexin knockout mice aged 9–12 months old. Similar changes in food intake and locomotor activity were observed in middle-aged male and female orexin knockout mice from the N10 generation (ESM Table 3).

Age-related deterioration in glucose tolerance in orexin knockout mice. Both orexin knockout (KO, black circles) and wild-type mice (WT, white circles) were maintained on a normal diet. a, b Changes in the body weight of male (a) and female (b) orexin knockout mice and the matched wild-type littermates from 4 to 36 weeks of age. Data are mean±SEM for four to seven mice per group. The inset shows epididymal fat pad weight (mg/g body weight) in 12-month-old mice. c–h Fasting and 120 min blood glucose levels measured during an OGTT in orexin knockout and wild-type mice. Male (c) and female (d) mice at 2 months of age, male (e) and female (f) mice at 6 months of age, and male (g) and female (h) mice at 9 months of age were fasted for 10 h, and then orally administered glucose (2 g/kg) at the 0-min timepoint. i, j The AUC for glucose concentrations during the 120 min OGTT in male (i) and female (j) wild-type (white bars) and orexin knockout (black bars) mice. The data are means ± SEM for three to 12 mice per group. a p < 0.05, b p < 0.01, c p = 0.068, d p = 0.085 vs wild-type mice
To investigate whether orexin deficiency affects glucose metabolism, orexin knockout mice at 2, 6 and 9 months of age underwent an OGTT. Fasting blood glucose levels were not significantly different between orexin knockout mice and wild-type mice at any age (Fig. 1c–h, at 0 min). After oral administration of glucose, no abnormalities in glucose tolerance were observed in either male (Fig. 1c) or female (Fig. 1d) 2-month-old orexin knockout mice, or in either male (Fig. 1e) or female (Fig. 1f) 6-month-old orexin knockout mice, compared with wild-type controls. However, in 9-month-old orexin knockout male mice, blood glucose levels were 1.2-fold and 1.3-fold higher at 15 and 120 min after glucose administration, respectively, compared with those in wild-type male mice (Fig. 1g), although the differences in the glucose AUCs did not reach significance (Fig. 1i). In addition, the glucose levels in the 9-month-old orexin knockout female mice were 1.3-fold higher at 60 and 120 min after glucose administration than those in wild-type female mice (Fig. 1h), and the glucose AUC was significantly increased (Fig. 1j). These results indicate that glucose tolerance was moderately impaired with age in orexin knockout mice.
In the insulin tolerance test, the response to insulin was not altered in either male (Fig. 2a) or female (Fig. 2b) 2-month-old orexin knockout mice, or in either male (Fig. 2c) or female (Fig. 2d) 6-month-old orexin knockout mice, compared with wild-type controls. In contrast, both male (Fig. 2e) and female (Fig. 2f) 9-month-old orexin knockout mice exhibited an apparent decrease in insulin sensitivity. The blood glucose levels in the orexin knockout male mice were 1.4-fold and 1.3-fold higher at 120 and 150 min after the insulin injection, respectively, compared with those in wild-type male mice (Fig. 2e), and the glucose AUC was significantly increased (Fig. 2g). In addition, the glucose levels in the knockout female mice were 1.9-fold and 2.4-fold higher at 120 and 150 min after the injection, respectively, compared with those in wild-type female mice (Fig. 2f), and a slight increase was observed in the glucose AUC (Fig. 2h). Thus, peripheral insulin sensitivity gradually reduced with age in orexin knockout mice.

Age-related development of insulin resistance in orexin knockout mice. Mice were maintained on normal diet. a–f Blood glucose levels measured during an insulin tolerance test in orexin knockout (KO, black circle) and wild-type mice (WT, white circle). Male (a) and female (b) mice at 2 months of age, male (c) and female (d) mice at 6 months of age, and male (e) and female (f) mice at 9 months of age were fasted for 10 h, and then intraperitoneally injected with insulin (1 U/kg) at the 0 min timepoint. Glucose levels are expressed as a percentage of the basal level. g, h The AUC for glucose concentrations during the 150 min insulin tolerance test in male (g) and female (h) wild-type (white bars) and orexin knockout (black bars) mice. The data are means ± SEM for three to eight mice per group. a p < 0.05, b p < 0.01, c p = 0.051 vs wild-type mice
Serum insulin levels were normal in 2-month-old orexin knockout mice (Fig. 3a), but were increased 2.7-fold in 6-month-old male orexin knockout mice, compared with wild-type controls (Fig. 3b). Furthermore, insulin levels were increased 5.3- and 2.6-fold in male and female 9-month-old orexin knockout mice, respectively (Fig. 3c). Serum leptin levels in male orexin knockout mice were not different from those in wild-type male mice at either 2 (Fig. 3d) or 9 months of age (Fig. 3e). In contrast, female orexin knockout mice showed a tendency towards increased serum leptin levels at 2 months of age (Fig. 3d), and showed a significant increase (2.2-fold) in leptin levels at 9 months of age (Fig. 3e). When the 2- to 3-month-old mice were maintained on a 60% high-fat diet, leptin levels were greatly elevated in both male and female wild-type mice, and tended to be further elevated in both the male and female orexin knockout mice (Fig. 3f). No difference was observed between the sexes with respect to leptin levels on the high-fat diet.

Age-related changes in serum levels of insulin and leptin in orexin knockout mice. After food deprivation for 12–14 h, blood samples were obtained from wild-type (WT) and orexin knockout mice (KO) maintained on a normal chow diet (NCD) or 60% high-fat diet (HFD) at the indicated ages. a–c Serum insulin levels in mice at 2 months (a), 6 months (b) and 9 months of age (c) on an NCD. d, e Serum leptin levels in mice at 2 months (d) and 9 months of age (e) on an NCD. f Serum leptin levels in mice at 2–3 months of age on 60% HFD for 5 weeks. The data are means ± SEM for four to 12 mice per group. * p < 0.05 vs wild-type mice
Orexin is a protection factor against diet-induced obesity and insulin resistance
When we challenged mice with a 30% high-fat diet from 3–24 weeks of age, male orexin knockout mice developed mild obesity (Fig. 4a), whereas female orexin knockout mice showed prominent weight gain compared with wild-type female mice (Fig. 4b). In the OGTT performed after high-fat dietary treatment for 6 months, blood glucose levels before and after glucose administration did not differ between male orexin knockout mice and wild-type littermates (Fig. 4c). In contrast, female orexin knockout mice exhibited significantly higher glucose levels before and 15 and 120 min after glucose administration compared with wild-type female mice (Fig. 4d). In the insulin tolerance test performed after 6 months on a high-fat diet, blood glucose levels were slightly higher in the orexin knockout male mice than those in wild-type male mice throughout the observation period, although the difference did not reach a significant level (Fig. 4e). However, in female orexin knockout mice, blood glucose levels at 60–150 min after the insulin injection were significantly higher than those in wild-type female mice (Fig. 4f).

Influence of a high-fat diet on glucose tolerance and insulin sensitivity in orexin knockout mice. Both orexin knockout (KO, black circles) and wild-type mice (WT, white circles) were maintained on 30% high-fat diet from 3 weeks to 6 months of age. a, b Changes in the body weight of male (a) and female (b) orexin knockout mice and the matched wild-type littermates. c, d Fasting and 120-min blood glucose levels measured during an OGTT. Male (c) and female (d) mice at 6 months of age were fasted for 10 h, and then orally administered glucose (2 g/kg) at the 0 min timepoint. e, f Blood glucose levels measured during an insulin tolerance test. Male (e) and female (f) mice at 6 months of age were fasted for 10 h, and then intraperitoneally injected with insulin (1 U/kg) at the 0 min timepoint. Glucose levels were expressed as a percentage of the basal level. The data are means ± SEM for four to 11 mice per group. *p < 0.05, **p < 0.01 vs wild-type mice
Orexin deficiency disrupts hypothalamic and peripheral insulin signalling
To investigate the influence of orexin deficiency on hypothalamic and peripheral insulin signalling, male orexin knockout mice fed a normal diet were used because, unlike the orexin knockout female mice, they did not exhibit obesity or serum leptin elevation. First, we analysed the level of phosphorylated Akt, a downstream molecule of phosphatidylinositol 3 (PI3)-kinase, in the hypothalamus of fasting orexin knockout mice at 2–3 months of age. Surprisingly, baseline levels of Akt phosphorylation at Ser473 in the knockout mice were approximately threefold higher than those in the wild-type mice, although hypothalamic levels of Akt were equivalent between these two groups of mice (Fig. 5a). The i.v. injection of insulin (4 U/kg) resulted in an approximately threefold increase in the levels of Akt phosphorylated at Ser473 in the hypothalamus of wild-type mice. However, the elevated basal levels of Akt phosphorylation in the knockout hypothalami were not increased, but, rather, were decreased by the insulin injection. Similarly, intracerebroventricular injection of insulin (10 mU) significantly increased the levels of hypothalamic phospho-Ser473-Akt in wild-type mice, but failed to affect the elevated basal levels of hypothalamic Akt phosphorylation in the orexin knockout mice (Fig. 5b). In contrast to the hypothalamus, the basal levels of phospho-Ser473-Akt in skeletal muscle were not different between wild-type and orexin knockout mice at 2–3 months of age, and i.v. injection of insulin (4 U/kg) effectively increased the phosphorylation levels in both wild-type and orexin knockout mice to a similar extent (Fig. 5c). Similar results were obtained in liver (Fig. 5d). These results indicate that insulin signalling is disrupted preferentially in the hypothalamus of orexin knockout mice at 2–3 months of age.

Impairment of both basal and insulin-induced Akt phosphorylation in the hypothalamus of young adult orexin knockout mice. After fasting overnight, young adult male mice (2–3 months of age) were injected with insulin, either intravenously (4 U/kg; a, c, d) or intracerebroventricularly (10 mU; b). Tissues were isolated from male wild-type (WT) and orexin knockout (KO) mice, 15 min after i.v. injection or 5 min after intracerebroventricular injection. a–d Levels of total Akt and Ser473-phosphorylated Akt in the hypothalamus (a and b), skeletal muscle (c) and liver (d), evaluated by Western blotting. Blots are representative of three to four independent experiments. Graphs show the relative level of Akt phosphorylation, calculated as the ratio of phosphorylated:total Akt in each sample. The data are the means ± SEM for three to four mice per group. *p < 0.05, **p < 0.01. p-, phosphorylated
When insulin was intravenously injected (4 U/kg) into 9-month-old wild-type mice, phosphorylation of Akt at Ser473 was efficiently induced in the hypothalamus (Fig. 6a), muscle (Fig. 6b) and liver (Fig. 6c). Elevation of basal phosphorylation of hypothalamic Akt (3.4-fold) was still observed in 9-month-old orexin knockout mice (Fig. 6a). Interestingly, the elevated basal levels in the hypothalamus were not affected by insulin injection. Furthermore, the insulin-induced increase in Akt phosphorylation observed in the wild-type mice was markedly reduced in muscle (Fig. 6b) and liver (Fig. 6c) of orexin knockout mice at this age. Similar results were observed for the phosphorylation of Akt at Thr308 (data not shown).

Impairment of insulin-induced Akt phosphorylation in both hypothalamus and peripheral tissues of middle-aged orexin knockout mice. After overnight fasting, middle-aged (9 months) male mice were intravenously injected with insulin (4 U/kg). Tissues were isolated from male wild-type (WT) and orexin knockout mice (KO) 15 min after the insulin injection. a–c Levels of total Akt and Ser473-phosphorylated Akt in the hypothalamus (a), skeletal muscle (b) and liver (c), evaluated by Western blotting. Blots are representative of three to four independent experiments. Graphs show the relative phosphorylation level of Akt, calculated as a ratio of phosphorylated:total Akt in each sample. The data are the means ± SEM for three to four mice per group. *p < 0.05, **p < 0.01. p-, phosphorylated
The insulin-induced tyrosine phosphorylation of insulin receptor β was reduced in the hypothalamus of 9-month-old orexin knockout mice compared with wild-type mice (Fig. 7a), whereas the phosphorylation levels in muscle (not shown) and liver (Fig. 7b) did not differ between wild-type and orexin knockout mice. In addition, there were no apparent changes in the levels of IRS-1 in the hypothalamus, muscle or liver of 9-month-old orexin knockout mice (Fig. 7c), IRS-2 in the hypothalamus of 2- to 3-month-old orexin knockout mice (data not shown) or glucose transporter GLUT4 in muscle of 9-month-old orexin knockout mice (Fig. 7c), compared with age-matched, wild-type mice. Phosphorylation of GSK3β, an Akt downstream target, also occurred in the hypothalamus, muscle and liver of 9-month-old wild-type mice in response to i.v. insulin injection (Fig. 7d–f). Basal levels of GSK3β phosphorylation in the hypothalamus of 9-month-old orexin knockout mice were 1.6-fold higher than those in the wild-type mice, and the elevated phosphorylation levels were not significantly altered by i.v. insulin injection (Fig. 7d). Furthermore, insulin failed to increase the levels of phosphorylated GSK3β in the muscle and liver of orexin knockout mice at this age (Fig. 7e,f).

Changes in the phosphorylation state of upstream and downstream molecules in the Akt signalling pathway in the hypothalamus and peripheral tissues of middle-aged orexin knockout mice. After overnight fasting, 9-month-old male mice were intravenously injected with insulin (4 U/kg). Tissues were isolated from male wild-type (WT) and orexin knockout mice (KO) 15 min after the insulin injection, and the lysate was directly subjected to Western blotting, except for the detection of tyrosine-phosphorylated insulin receptor β (IRβ). a, b Levels of total and tyrosine-phosphorylated IRβ in the hypothalamus (a) and liver (b). To detect the phosphorylated IRβ, the lysate was immunoprecipitated with anti-IRβ antibody and then immunoblotted with anti-PY99 antibody. c Levels of IRS-1 in the hypothalamus, muscle and liver, and GLUT4 in muscle. d–f Levels of total GSK3β and GSK3β phosphorylated at Ser9 in the hypothalamus (d), skeletal muscle (e) and liver (f). g, h Levels of total and phosphorylated p42 MAP kinase in the hypothalamus (g) and skeletal muscle (h). i Levels of total AMP kinase (AMPK) and AMPKα phosphorylated at Thr172 in the hypothalamus. Blots are representative of three to five independent experiments. Graphs show the relative phosphorylation level of each protein, calculated as the ratio of phosphorylated to total protein level. The data are the means ± SEM for three to five mice per group. *p < 0.05, **p < 0.01. p-, phosphorylated
Levels of p44/p42 MAP kinase phosphorylation in the hypothalamus were not altered by i.v. insulin injection in either wild-type or orexin knockout mice at 9 months of age (Fig. 7g). Although basal levels of p44/p42 MAP kinase phosphorylation were elevated in skeletal muscle of orexin knockout mice, an increase in MAP kinase phosphorylation was induced in muscle of both wild-type and orexin knockout mice by insulin injection (Fig. 7h).
Basal levels of AMP kinase α phosphorylation in orexin knockout mice were significantly lower than those of wild-type mice (Fig. 7i), indicating that hypothalamic AMP kinase activity was reduced by orexin deficiency. These results were in agreement with the reduced food intake in orexin knockout mice, since the decreased AMP kinase activity in the hypothalamus is known to inhibit food intake [27].
Finally, we examined the influence of high-fat feeding on insulin signalling in male and female wild-type and orexin knockout mice. When the 2- to 3-month-old mice were fed a 60% high-fat diet for 6 weeks, the body weight gain was more evident in female orexin knockout mice than wild-type female mice, whereas the weight gain was comparable for the two groups of male mice (data not shown). After insulin stimulation, the wild-type and orexin knockout male mice exhibited a similar increase in levels of Akt phosphorylation in muscle (ESM Fig. 1a) and liver (ESM Fig. 1b), whereas no significant increases in phosphorylation levels were observed in the hypothalamus (data not shown). Similar results were obtained in the female wild-type and orexin knockout mice (ESM Fig. 1c,d). These results indicate that there is no difference between the sexes in terms of insulin-induced phosphorylation of Akt in orexin knockout mice fed a high-fat diet.
Discussion
Orexin regulates motivated behaviours, such as arousal, feeding and reward-seeking, and has important effects on metabolic, autonomic and endocrine functions [28–30]. The findings that orexin-A increases oxygen consumption and body temperature even in anaesthetised rats [31], and that orexin neuron-deficient mice display reduced energy expenditure that is independent of sleep/wake, locomotion and food intake [32] indicate that orexin-induced increase in energy metabolism is not simply due to increased wakefulness and physical activity. In the present study, we investigated the involvement of orexin on glucose metabolism using orexin knockout mice, and found that orexin is essential for the maintenance of normal insulin sensitivity with increasing age, although the secondary influences of reduced chronic physical activity and energy expenditure on insulin resistance with age cannot be excluded using this animal model.
Insulin resistance is a major factor in the pathogenesis of type 2 diabetes, which occurs during the ageing process and results in impaired glucose metabolism [33, 34]. We observed that male orexin knockout mice exhibited impaired insulin sensitivity and hyperinsulinaemia at 9 months but not at 2–6 months of age. Glucose tolerance was not markedly impaired in middle-aged male orexin knockout mice, probably because insulin resistance could be compensated by hyperinsulinaemia to maintain glucose tolerance. Female orexin knockout mice exhibited impaired glucose tolerance, slight insulin resistance and moderate hyperinsulinaemia at 9 months but not at 2–6 months of age. Moreover, late-onset mild obesity was observed exclusively in female orexin knockout mice, consistent with previous reports [17, 35]. These results indicate that male orexin knockout mice developed age-related insulin resistance without obesity, whereas insulin resistance and some other factors related to obesity may synergistically cause abnormal glucose tolerance in female orexin knockout mice.
Both orexin receptors and insulin receptors are abundant in the hypothalamus, and promote peripheral energy metabolism, despite having opposite effects on food intake [23, 36, 37]. Brain insulin receptor deficiency causes obesity and mild insulin resistance in mice [38], similar to that seen in animals with orexin deficiency. Insulin receptor signalling is mediated by two main pathways: the PI3-kinase/Akt and MAP kinase pathway [20, 39]. The hypothalamic PI3-kinase/Akt pathway mediates the effect of insulin on food intake and energy expenditure via forkhead transcription factor Foxo1 [40], whereas insulin-dependent activation of hypothalamic MAP kinase does not have any effect on glucose homeostasis [23]. We found that basal levels of Akt phosphorylation were elevated in the hypothalamus of orexin knockout mice, and central and peripheral injection of insulin failed to increase the phosphorylation levels. These results indicate that hypothalamic insulin signalling is disrupted by orexin deficiency. Thus, orexin and insulin appear to exert cooperative actions on energy metabolism in the hypothalamus. Basal levels of Akt in the hypothalamus of wild-type mice were increased by intracerebroventricular injection of 4 μmol/l orexin at a rate of 0.25 μl/h, for a total of 14 days, using an osmotic minipump. The elevated levels of Akt phosphorylation in the hypothalamus of orexin knockout mice were not reversed, but, rather, enhanced by orexin administration (data not shown). It is possible that longer term or more finely tuned treatment with orexin, mimicking the physiological secretion pattern, might be necessary to rescue the abnormal hypothalamic Akt signalling in orexin deficiency.
Orexin neurons are known to discharge during active waking [41], and the excitability is inhibited by glucose [1, 4, 6]. In addition, changes in blood glucose levels negatively correlate with expression of the gene encoding prepro-orexin in the mouse hypothalamus [11]. A recent study demonstrated that blood glucose is a crucial for the regulation of Akt phosphorylation levels in the cerebral cortex and hippocampus of mice, reporting that phosphorylation levels are lowered in the fasting state and elevated after glucose administration in non-diabetic mice, and that phosphorylation levels are elevated in streptozotocin-induced diabetic mice [42]. We therefore anticipate that the basal level of Akt phosphorylation is extensively elevated in the brain, including the hypothalamus, under conditions where expression of the gene for prepro-orexin is reduced or lost. The glucosensing machinery of the orexin neurons may be at least partly involved in the conversion of hyperglycaemic signals into Akt phosphorylation through an as yet unknown mechanism.
Although hypothalamic insulin action appears to play a crucial role in the regulation of peripheral insulin actions, in 2- to 3-month-old male orexin knockout mice, Akt signalling is perturbed in the hypothalamus but normal in the peripheral tissues. We consider that some compensatory mechanism(s) may serve to maintain normal insulin sensitivity and glucose metabolism in the periphery. Alternatively, the possibly subtle changes in Akt signalling in the peripheral tissues of orexin knockout mice at this age may be undetectable under the conditions used in our experiments. In contrast, long-term (9 months) deficiency of orexin caused greater disruption of hypothalamic insulin signalling, which seemed to indirectly produce more severe defects in the peripheral actions of insulin. Our views are supported by the fact that the young adult (2-month-old) male orexin knockout mice exhibited a slight increase in serum insulin levels, whereas the middle-aged (9-month-old) knockout male mice exhibited marked hyperinsulinaemia accompanying insulin resistance. Given that central orexin levels are reduced by hyperglycaemia associated with peripheral insulin resistance, a vicious cycle appears to be formed between peripheral and hypothalamic insulin resistance through lowering orexin levels (Fig. 8). We suggest that in response to orexin deficiency, basal levels of Akt phosphorylation in the hypothalamus are abnormally increased throughout the ageing process, and the long-term phosphorylation disrupts the hypothalamic insulin action regulating peripheral insulin sensitivity. The reduced phosphorylation and activation of the hypothalamic insulin receptor may partly explain this mechanism. Since lack of Akt2, but not Akt1 or Akt3, in both central and peripheral tissues, leads to metabolic abnormalities in mice [43], the reduction of insulin-induced Akt2 activity might be involved in the vicious cycle, although Akt isoform-specific functions in the hypothalamus have yet to be clarified.

A putative model describing how orexin deficiency causes peripheral insulin resistance with age. Hypothalamic insulin action via the Akt signalling pathway helps to maintain peripheral insulin sensitivity for glucose metabolism. Under conditions of orexin deficiency, such as prepro-orexin gene ablation and long-term hyperglycaemia, basal levels of hypothalamic Akt phosphorylation are increased throughout the ageing process, and the long-term phosphorylation disrupts hypothalamic insulin action. Thus, insulin resistance and associated hyperglycaemia could elicit a vicious cycle of worsening insulin resistance with ageing by reducing orexin levels
A high-fat diet is known to increase the risk of insulin resistance. When fed a high-fat diet, obesity was more evident in female orexin knockout mice than wild-type female mice, and was associated with impaired fasting glucose, severe glucose intolerance and insulin resistance. In contrast, no such differences were observed between the orexin knockout male mice and the wild-type male mice. Although the biological basis of the difference between the sexes remains unclear, the present results indicate that alteration of Akt phosphorylation is not the key factor. We anticipate that leptin plays a pivotal role in this regard, because diet-induced obesity leads to leptin resistance that could further exacerbate insulin resistance [44, 45], and because brain uptake of leptin decreases in female and elderly humans [46]. In fact, serum leptin levels were elevated exclusively in middle-aged orexin knockout female mice fed a normal chow diet, consistent with previous observation in female mice deficient in orexin neurons [35]. Further study will be required to determine the influence of orexin deficiency in leptin signalling, especially in females.
In conclusion, the present results demonstrate that orexin is an essential factor for maintaining hypothalamic insulin signalling, which is likely to be responsible for preventing the development of peripheral insulin resistance with advancing age. This may explain why the incidence of type 2 diabetes increases in orexin-deficient human narcolepsy without severe obesity. Furthermore, the expression of the gene for prepro-orexin is downregulated in diabetic ob/ob and db/db mice [47], and the downregulation is shown to be due to hyperglycaemia in these animals [4]. These suggest that diabetic hyperglycaemia reduces the expression of the gene for prepro-orexin in the hypothalamus, and thereby further worsens the insulin resistance. Therefore, the orexin receptor that controls hypothalamic insulin signalling may be a new target to treat hyperglycaemia in patients with type 2 diabetes.
Abbreviations
- GSK:
-
glycogen synthase kinase
- MAP:
-
mitogen-activated protein
- PI:
-
phosphatidylinositol
References
Sakurai T (2007) The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci 8:171–181
de Lecea L, Kilduff TS, Peyron C et al (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A 95:322–327
Sakurai T, Amemiya A, Ishii M et al (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585
Yamanaka A, Beuckmann CT, Willie JT et al (2003) Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38:701–713
Burdakov D, Gerasimenko O, Verkhratsky A (2005) Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci 25:2429–2433
Burdakov D, Jensen LT, Alexopoulos H et al (2006) Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron 50:711–722
Moriguchi T, Sakurai T, Nambu T, Yanagisawa M, Goto K (1999) Neurons containing orexin in the lateral hypothalamic area of the adult rat brain are activated by insulin-induced acute hypoglycemia. Neurosci Lett 264:101–104
Griffond B, Risold PY, Jacquemard C, Colard C, Fellmann D (1999) Insulin-induced hypoglycemia increases preprohypocretin (orexin) mRNA in the rat lateral hypothalamic area. Neurosci Lett 262:77–80
VanItallie TB (2006) Sleep and energy balance: interactive homeostatic systems. Metabolism 55:S30–S35
Thannickal TC, Moore RY, Nienhuis R et al (2000) Reduced number of hypocretin neurons in human narcolepsy. Neuron 27:469–474
Sakurai T (2005) Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med Rev 9:231–241
Honda Y, Doi Y, Ninomiya R, Ninomiya C (1986) Increased frequency of non-insulin-dependent diabetes mellitus among narcoleptic patients. Sleep 9:254–259
Lammers GJ, Pijl H, Iestra J, Langius JA, Buunk G, Meinders AE (1996) Spontaneous food choice in narcolepsy. Sleep 19:75–76
Schuld A, Hebebrand J, Geller F, Pollmacher T (2000) Increased body-mass index in patients with narcolepsy. Lancet 355:1274–1275
Nishino S, Ripley B, Overeem S et al (2001) Low cerebrospinal fluid hypocretin (orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol 50:381–388
Hara J, Beuckmann CT, Nambu T et al (2001) Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30:345–354
Hara J, Yanagisawa M, Sakurai T (2005) Difference in obesity phenotype between orexin-knockout mice and orexin neuron-deficient mice with same genetic background and environmental conditions. Neurosci Lett 380:239–242
Burdakov D, Alexopoulos H (2005) Metabolic state signalling through central hypocretin/orexin neurons. J Cell Mol Med 9:795–803
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96
Gerozissis K (2004) Brain insulin and feeding: a bi-directional communication. Eur J Pharmacol 490:59–70
Plum L, Belgardt BF, Brüning JC (2006) Central insulin action in energy and glucose homeostasis. J Clin Invest 116:1761–1766
Prodi E, Obici S (2006) Minireview: the brain as a molecular target for diabetic therapy. Endocrinology 147:2664–2669
Tsuneki H, Sugihara Y, Honda R, Wada T, Sasaoka T, Kimura I (2002) Reduction of blood glucose level by orexins in fasting normal and streptozotocin-diabetic mice. Eur J Pharmacol 448:245–252
Chemelli RM, Willie JT, Sinton CM et al (1999) Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98:437–451
Wada T, Naito M, Kenmochi H, Tsuneki H, Sasaoka T (2007) Chronic nicotine exposure enhances insulin-induced mitogenic signaling via up-regulation of α7 nicotinic receptors in isolated rat aortic smooth muscle cells. Endocrinology 148:790–799
Minokoshi Y, Alquier T, Furukawa N et al (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428:569–574
Willie JT, Chemelli RM, Sinton CM, Yanagisawa M (2001) To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci 24:429–458
Carr D, Kalivas PW (2006) Orexin: a gatekeeper of addiction. Nat Med 12:274–276
Spinazzi R, Andreis PG, Rossi GP, Nussdorfer GG (2006) Orexins in the regulation of the hypothalamic–pituitary–adrenal axis. Pharmacol Rev 58:46–57
Wang J, Osaka T, Inoue S (2001) Energy expenditure by intracerebroventricular administration of orexin to anesthetized rats. Neurosci Lett 315:49–52
Zhang S, Zeitzer JM, Sakurai T, Nishino S, Mignot E (2007) Sleep/wake fragmentation disrupts metabolism in a mouse model of narcolepsy. J Physiol 581:649–663
Pessin JE, Saltiel AR (2000) Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest 106:165–169
Cook SA, Aitman T, Naoumova RP (2005) Therapy insight: heart disease and the insulin-resistant patient. Nat Clin Pract Cardiovasc Med 2:252–260
Fujiki N, Yoshida Y, Zhang S, Sakurai T, Yanagisawa M, Nishino S (2006) Sex difference in body weight gain and leptin signaling in hypocretin/orexin deficient mouse models. Peptides 27:2326–2331
de la Monte SM, Wands JR (2005) Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis 7:45–61
Sakurai T (2006) Roles of orexins and orexin receptors in central regulation of feeding behavior and energy homeostasis. CNS Neurol Disord Drug Targets 5:313–325
Brüning JC, Gautam D, Burks DJ et al (2000) Role of brain insulin receptor in control of body weight and reproduction. Science 289:2122–2125
Zhao WQ, Chen H, Quon MJ, Alkon DL (2004) Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol 490:71–81
Kim MS, Pak YK, Jang PG et al (2006) Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci 9:901–906
Lee MG, Hassani OK, Jones BE (2005) Discharge of identified orexin/hypocretin neurons across the sleep–waking cycle. J Neurosci 25:6716–6720
Clodfelder-Miller B, De Sarno P, Zmijewska AA, Song L, Jope RS (2005) Physiological and pathological changes in glucose regulate brain Akt and glycogen synthase kinase-3. J Biol Chem 280:39723–39731
Dummler B, Tschopp O, Hynx D et al (2006) Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol 26:8042–8051
El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS (2000) Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest 105:1827–1832
Frühbeck G (2006) Intracellular signalling pathways activated by leptin. Biochem J 393:7–20
Arnulf I, Lin L, Zhang J et al (2006) CSF vs serum leptin in narcolepsy: is there an effect of hypocretin deficiency? Sleep 29:1017–1024
Yamamoto Y, Ueta Y, Date Y et al (1999) Down regulation of the prepro-orexin gene expression in genetically obese mice. Mol Brain Res 65:14–22
Acknowledgements
We thank S. Kobayashi, T. Uwano and H. Nishijo (University of Toyama) for their technical assistance. This study was supported in part by Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (to H. Tsuneki) and the Japan Diabetes Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
The influence of high-fat feeding on Akt signalling in male and female wild-type and orexin knockout mice. Young adult mice (2–3 months of age) were maintained on a 60% high-fat diet (HFD) for 6 weeks. After overnight fasting, the mice were intravenously injected with insulin (4 U/kg). Tissues were isolated from wild-type (WT) and orexin knockout (KO) mice 15 min after the insulin injection, and subjected to Western blotting. a–d The phosphorylation levels of Akt at Ser473 in skeletal muscle (a) and liver (b) of male mice, and in skeletal muscle (c) and liver (d) of female mice. Blots are representative of three independent experiments. Data are means ± SEM for three mice per group. *p < 0.05 (DOC 54.6 kb)
Table S1
Sequences of the PCR primers used to genotype the offspring of heterozygous (+/−) mice (PDF 13.2 kb)
Table S2
Phenotype characteristics of 9- to 12-month-old female orexin knockout mice from the N5 generation (PDF 16.5 kb)
Table S3
Feeding behavior and locomotor activity of 1-year-old orexin knockout mice from the N10 generation (PDF 17.0 kb)
Rights and permissions
About this article
Cite this article
Tsuneki, H., Murata, S., Anzawa, Y. et al. Age-related insulin resistance in hypothalamus and peripheral tissues of orexin knockout mice. Diabetologia 51, 657–667 (2008). https://doi.org/10.1007/s00125-008-0929-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-0929-8