
Abstract
Aims/hypothesis
The human placenta is a complex organ situated at the interface between mother and foetus that separates maternal from foetal blood. The placental surfaces exposed to the two bloodstreams are different, i.e. trophoblasts and endothelial cells are in contact with the maternal and foetal circulation, respectively. Both cell types produce high insulin receptor levels. The aim of the present study was to test the hypothesis that spatio-temporal changes in insulin receptor expression in trophoblasts from first trimester to the endothelium at term shift the control of insulin-dependent processes from mother to foetus.
Methods
Global microarray analysis of primary trophoblasts from first trimester and term human placentas and endothelial cells from term human placentas cultured under hyperinsulinaemic and control conditions identified different sets of regulated genes in trophoblasts and endothelial cells.
Results
Insulin effects on placental gene expression underwent developmental changes from trophoblasts in the first trimester to endothelial cells at term that were paralleled by changes in levels of activated insulin receptors. The changes in gene regulation were both quantitative (i.e. magnitude of effect) and qualitative (i.e. specific genes affected and direction of regulation).
Conclusions/interpretation
This spatio-temporal shift in insulin sensitivity throughout pregnancy allows maternal and foetal insulin to regulate different processes within the placenta at different gestational stages, facilitated by compartmentalisation of the insulin response. Thus, by altering the levels and function of insulin receptors in space and time, control of insulin-dependent processes in the human placenta will change from mother to foetus throughout gestation. This will be of particular interest in conditions associated with altered maternal or foetal insulin levels, i.e. diabetes mellitus or intrauterine growth restriction.
Similar content being viewed by others

Introduction
The mechanisms by which the metabolic and hormonal environment regulate the growth of the foetus and may cause foetal overgrowth in diabetes are poorly understood. Intrauterine maldevelopment leads to foetuses with a birthweight inappropriate for their gestational age (i.e. overweight or underweight), who are at a higher risk of developing obesity and metabolic disorders later in life [1]. Animal studies and observations in humans clearly showed that in addition to genetics, the intrauterine environment also has a strong influence on foetal growth and development [2–4].
The human placenta is a foetal organ that develops within the uterus. Its growth precedes that of the foetus proper. As a complex organ, it is composed of a variety of different cells. Owing to its position at the interface between mother and foetus, the placenta separates maternal and foetal blood. The surfaces exposed to the two bloodstreams are different: the maternal circulation is in contact with the trophoblasts whereas the foetal circulation is in contact with the placental endothelium. Both cell types express high levels of insulin receptors [5].
Endocrine or metabolic changes in either circulation may affect the placenta, and the specific effect may differ depending on the specialised function of the cells [6, 7]. Maternal diabetes mellitus is associated with changes in the maternal and foetal circulation. In insulin-treated gestational diabetes (GDM), maternal hyperinsulinism is a characteristic metabolic feature. Hyperinsulinism is also found in the foetal circulation as a result of the stimulation of the foetal pancreas by elevated levels of insulin secretagogues [8]. Therefore, diabetes in pregnancy represents a condition in which insulin acts on two different surfaces of one tissue, i.e. the human placenta.
Insulin can influence cells in a diverse manner. It can modulate intracellular metabolism by altering the activity or intracellular location of various enzymes, and it can regulate gene expression. More than 150 insulin-responsive genes are known [9–11], and these encode proteins related to various cellular functions. The specific insulin response depends on the type and stage of differentiation of the cell.
Maternal and foetal insulin may induce opposing effects and may even discordantly alter placental function if the responses are not controlled and coordinated. One possible mechanism of control involves the separation of effects in time (i.e. gestational age) and space (i.e. the two surfaces), and the aim of this study was to investigate this mechanism.
The availability of methods to isolate primary cells from both surfaces from the human placenta, i.e. trophoblasts and endothelial cells (ECs), enabled us to analyse in vitro the insulin-induced effects separated by space. In addition, trophoblasts can be obtained from the beginning and the end of gestation and, hence, be used to analyse the effect of time. In the first trimester, placental ECs are very sparse and do not produce insulin receptors at levels detectable by immunohistochemistry [5] and, hence, may not compete for insulin effects in the placenta.
In the present study we used global gene expression to investigate whether there is a shift in the sensitivity of insulin-mediated gene expression from the maternal to the foetal surface of the human placenta over the course of pregnancy.
Materials and methods
Isolation of cells
Trophoblasts (TTs) and ECs were isolated from term placentas after uncomplicated pregnancy and vaginal delivery following a standard protocol [12, 13]. First-trimester trophoblasts (FTs) were isolated from placental tissue after pregnancy terminations for psychosocial reasons (seventh to tenth week of pregnancy) as described previously [13].
Characterisation and culture of human trophoblasts and ECs
All cell preparations were subjected to rigorous immunocytochemical characterisation [12–14]. Trophoblasts were tested for viability by measuring secreted human chorionic gonadotropin hormone (hCG) levels using a commercially available assay (Dade Behring, Deerfield, IL, USA). Only preparations of ≥99% purity and the characteristic kinetics of hCG secretion [15] were used. Prior to the addition of insulin, trophoblasts were cultured for 24 h in DMEM (GIBCO, Paisley, UK) with 10% FCS, 100 mg/ml streptomycin and 100 IU/ml penicillin (GIBCO), to allow adaptation to the in vitro conditions.
ECs were characterised by internalisation of acetylated LDL (Biomedical Technologies, Stoughton, MA, USA) [12]. They were cultured on 1% (v/v) gelatin-coated plates using Endothelial Basal Medium (Clonetics, Cambrex, Walkersville, MD, USA) supplemented with the EGM-MV BulletKit (Clonetics) until 70% confluency, at which time insulin was added.
Each cell preparation was divided into two subpopulations, which were cultured in appropriate medium under normoinsulinaemic (9 pmol/l; 1.5 μU/ml) or hyperinsulinaemic (1 nmol/l; 167 μU/ml) conditions for 16 h. Insulin was from ICN Biomedicals (Aurora, OH, USA), and media concentrations of insulin were measured by RIA (IMP Biomedicals, Doornveld, Belgium).
RNA isolation and processing
RNA was isolated using Trizol (MRC, Cincinnati, OH, USA) followed by quality assessment using a bioanalyser (Agilent, Palo Alto, CA, USA). Experimental procedures and data analysis followed recommended standards [16]. Total RNA from ten preparations per cell type, isolated from different placentas, was pooled [17]. Using 5 μg of pooled RNA, cDNA was synthesised (SuperScript Double-Stranded cDNA Synthesis Kit, Invitrogen, Carlsbad, CA, USA), transcribed in vitro (RNA Transcript Labeling Kit, Enzo diagnostics, Farmingdale, NY, USA) and then fragmented. To test the quality of the cRNA, it was hybridised against Test-3 arrays (Affymetrix, Santa Clara, CA, USA). As samples passed the quality criteria (bioC, bioD and cre were present, the 3′:5′ ratio of the polyA controls was <3), the cRNAs were hybridised against Affymetrix HU133A-chips. RNA preparation and hybridisation was performed according to the Affymetrix user manual.
Data analysis
Raw data were normalised globally and processed with Microarray Suite, version 5.0 (Affymetrix) and Data Mining Tool (Affymetrix) software. Genes that met the following three criteria were classed as being differentially expressed: (1) fold change ≥1.5 or ≤−1.5; (2) change in p value ≥0.992 or ≤0.008; and (3) at least one signal intensity (control or treatment) >100. Annotations were obtained from NetAffx (available at http://www.affymetrix.com, last accessed in October 2005). Regulated genes selected as above were clustered manually in Microsoft Excel according to the encoded proteins' putative function using database searches on PubMed and OMIM (available at http://www.ncbi.nlm.nih.gov, last accessed in October 2005).
Semiquantitative RT-PCR
Primers targeting RPL30 (CCT AAG GCA GGA AGA TGG TG; CAG TCT GTT CTG GCA TGC TT), CYP51A1 (AAC CTC TTG TCC ATG CTG CT; GCA TCA CTC CCC AGA AGG TA), EFNB2 (CTG CTG GAT CAA CCA GGA AT; CCG GTA CTT CAG CAA GAG GA), EZH1 (CTG GAC TGT GGG TTC CAA AT, CTG AAG CTG GGA CAG GAG AC), PTBP1 (AAG GTC CTG TTT TCC AGC AA; CCC TAG ATG GTG GAC TTG GA) and VIL2 (GCC AAG TTT GGG GAC TAC AA; TTT CCA GGT CCT GAG CAA TC) were designed using the public web-page Primer3 (available at http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi, last accessed in October 2005) and purchased from MWG Biotech (Ebersberg, Germany). The primer pairs hybridised within the target sequence of the HU133A-chip and, wherever possible, included splicing sites within the amplicon. The transcription of RPL30 (which encodes ribosomal protein L30) was used as an internal control [18]. For validation, RNA from newly isolated and insulin-treated cells was used following the protocol for the microarray analysis. Total RNA (100 ng) was used in the One-Step RT-PCR Kit from Quiagen (Hilden, Germany) according to the manufacturer's instructions. The optimum cycle number was determined for each primer pair. PCR products were separated by electrophoresis through agarose gels (2%), documented with the Eagle-Eye system (Stratagene, La Jolla, CA, USA) and quantified using AlphaDigiDoc 1,000 (Alpha Innotech, San Leandro, CA, USA) software.
Real-time PCR
The cDNA was synthesised from 500 ng RNA according to the protocol used for the microarray experiment. Real-time PCR was performed with Assay-on-Demand Gene Expression Products from Applied Biosystems for the relevant genes using TaqMan and the ABI Prism 5,700 Sequence Detection System. Data were analysed according to the \(2^{{ - \Delta \Delta C}} {\text{T}}\) method [19]. Levels of RPL30 were measured as an internal control [18].
Western blot for insulin receptors and phosphorylated insulin receptors
Cells were serum starved overnight. Insulin was added to a final concentration of 1 μmol/l (167 mU/ml) for 4 min. No insulin was added to the controls. Cells were lysed in buffer containing 0.01 mol/l Tris pH 7.4, 1% SDS, 1 mmol/l sodium orthovanadate and Complete Protease Inhibitor (Roche, Mannheim, Germany) mixed with an equal volume of Laemmli sample buffer (Sigma, St Louis, MO, USA). Prior to electrophoresis, samples were centrifuged and boiled for 5 min at 99°C. Equal amounts of protein, determined by the method of Lowry et al. [20], were used for SDS-PAGE on an 8–18% gradient gel (ExcelGel; Pharmacia, Uppsala, Sweden). After semi-dry electroblotting (Excel system; Pharmacia) according to the manufacturer’s instructions, membranes were blocked for 1 h with 5% (w/v) non-fat dry milk (BioRad, Hercules, CA, USA) and 0.1% (v/v) Tween-20 (Sigma) in 0.14 mol/l Tris-buffered saline pH 7.3 at room temperature. This solution was used for subsequent washings and as a diluent for the antibodies. The membranes were incubated with the antibodies against insulin receptors (1:300 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and Tyr1162/Tyr1163-phosphorylated insulin receptors (Calbiochem, San Diego, CA, USA) overnight at 4°C. After washing, the membranes were incubated with the secondary antibody (1:1,000 dilution; BioRad) for 1 h at room temperature. Immunolabelling was visualised using the SuperSignal CL-HRP Substrate System (Pierce, Rockford, IL, USA). Membranes were exposed to Hyperfilm (Amersham, Chalfont St Giles, UK) and than analysed using a digital camera and the AlphaDigiDoc 1,000 software.
Results
Microarray analysis
Of the 22,215 transcripts analysed, the levels of 236 and 146 transcripts were either increased or decreased by insulin by a factor >1.5 in FTs and ECs, respectively (Fig. 1). Only six differentially expressed genes were identified in TTs (Fig. 1). In trophoblasts, most genes were downregulated (91% in FTs; 83% in TTs), whereas, at term, 45% of the differentially expressed genes in ECs were upregulated (Fig. 1). This spatio-temporal change in gene response was also seen when more stringent cut-off levels (>2-fold change, >2.5-fold change) were applied (not shown). On average, insulin induced an approximately 2-fold change in gene expression levels (for the complete list of regulated transcripts see Electronic supplementary material).

Number of genes upregulated or downregulated by >1.5-fold by 1 nmol/l insulin as compared with 9 pmol/l insulin in FTs, TTs and ECs. The proportion of downregulated genes is shown within the bars. The open bars represent the number of upregulated genes, the closed bars represent the number of downregulated genes
Validation of the microarray experiment
The insulin-induced changes in gene expression detected by the microarray experiment were verified by semiquantitative RT-PCR or real-time PCR (Table 1). The particular genes verified were selected such that diverse cellular functions and all placental cell types analysed were covered. Although the differential expression of the selected genes was verified most of the changes were smaller than those detected by the microarray analysis.
Functional clustering of the regulated genes
The insulin-regulated genes had very diverse cellular functions. The greatest proportion of genes were related to DNA and transcription, signal transduction and metabolism in FTs, and to DNA and transcription, signal transduction and growth factors/cell cycle/apoptosis in ECs (Fig. 2). The low number of regulated genes in TTs did not allow meaningful clustering. Although the pattern of regulated functional clusters was similar in all cell types, only a few genes were regulated by insulin in more than one cell type (Fig. 3). No insulin-sensitive gene was found to be common to FTs and TTs. A number of differentially regulated genes in the placenta were also found in other tissues by other authors (Tables 2 and 3), but the direction of regulation differed in several cases.

Functional clusters of regulated genes under hyperinsulinaemia. The bar length represents the number of genes within each cluster. ECM, extracellular matrix

Venn diagram, showing the number of genes that were differentially expressed in FT and EC. The genes regulated by insulin (1 nmol/l) in more than one cell type are listed in the right panel and printed in bold if they are regulated in same direction in both cell types. The genes regulated only in TT are listed in the left panel
Insulin receptor expression and phosphorylation
Western blot analysis demonstrated high insulin receptor levels in FTs. TTs produced 70% fewer (p<0.05) insulin receptors. At term, ECs were the predominant site of insulin receptor expression, with about twice as many receptors as the TTs (Fig. 4). In all cells the receptors could be autophosphorylated by insulin within 4 min (Fig. 4a). Activation by insulin was strongest in FTs and lowest in TTs (Fig. 4c). Activated receptor normalised to total receptor was similar in all cell types (not shown). According to the microarray analysis, insulin treatment did not alter levels of INSR mRNA in the different cell types.

Insulin receptor production and activation. a Representative western blot of proteins from FTs, TTs and ECs probed with antibodies against total insulin receptor and phosphorylated insulin receptor (PY-1162, PY-1163) b Densitometric quantification of total insulin receptor. c Difference in insulin receptor phosphorylation of insulin-stimulated vs control cells. Data are presented as means±SD; C, control; I, insulin-treated (4 min); IR, insulin receptor
Discussion
The hypothesis of a change in placental control from mother to foetus is of significance against the background of hyperinsulinism in the maternal and foetal circulation in diabetic pregnancies. Based on this we investigated the effects of hyperinsulinism on distinct placental cells that may contribute to changes in placental tissue in diabetes. The experimental conditions chosen mimicked the in vivo situation as closely as possible: cells were not serum starved prior to hyperinsulinaemia and a low, yet pathophysiological, insulin concentration was used. There was a 100-fold difference in the insulin concentration between the control and hyperinsulinaemic conditions. The 1 nmol/l (167 μU/ml) insulin concentration was chosen as to reflect the higher range of maternal insulin levels seen in response to hyperglycaemia in GDM [21] or in the cord blood of type 1 diabetic pregnancies [22]. It also represents the upper limit of fasting insulin levels in type 1 diabetic mothers [23]. The exposure time of 16 h was chosen in order to allow transcription to occur, but limiting secondary effects. The fact that these conditions are close to those in vivo may explain the small magnitude of alterations in gene expression observed in vitro. In all three cell types, most of the changes (FT 89%, TT 83%, EC 88%) in gene expression were not greater than a factor of 2.5. These values are similar to those reported by another microarray study in an in vivo situation that analysed human skeletal muscle tissue during a hyperinsulinaemic clamp [24].
We identified 371 insulin-responsive genes that were specifically regulated in one compartment of the placenta. Some of the genes had also been identified in other microarray studies, which used different cell models, stimulation and analysis modalities [24–28], or by other methods [9, 10], thus representing a robust insulin response common to many cell types.
The insulin-responsive genes encoded proteins involved in diverse cellular functions extending beyond the classical functions of insulin (i.e. the transport and metabolism of glucose). Only in some cases were genes regulated in more than one cell type (Fig. 3), but the general pattern of regulated functional clusters was similar: most genes were related to DNA and transcription and signal transduction. Consistent with other studies [24, 28], a large number of genes assigned to metabolism were differentially expressed (Fig. 2). The proportion of genes in the ‘metabolism’ cluster was higher in the FTs, whereas the proportion of genes in the ‘growth factors/cell cycle/apoptosis’ cluster was higher in the ECs. Therefore, one could hypothesise that insulin affects metabolism to a greater extent in FTs, whereas the cell cycle is more strongly affected in ECs. Nevertheless, one has to keep in mind the difficulty of elucidating functions from a set of regulated genes. Literature describing the effects of insulin in FTs or ECs remains to be published; however, we are currently testing the above hypothesis in our laboratory. Similar to the changes observed in the placental tissue in diabetes [29], a great proportion (FT 58%; EC 55%) of the genes in the metabolism cluster were involved in various aspects of lipid biosynthesis and fatty acid metabolism. Also in line with gene expression changes in placental tissue from diabetic women was the absence of changes in genes related to glucose transport and metabolism. This may in part explain the lack of an effect of insulin on placental glucose transport and metabolism [30]. Levels of mRNA for PCK2, a classical insulin-regulated gene encoding the key enzyme in gluconeogenesis, were below the microarray detection limit in all cell types, which is in line with the absence of placental gluconeogenesis [30].
Comparison of insulin-regulated genes in isolated TTs and term ECs with genes expressed differentially in term placental tissue obtained from insulin-treated GDM women using microarrays similar to those used in this study [29] only identified a few genes that were regulated under both in vivo and in vitro conditions (Table 4). There are several possible reasons for the limited number detected: the influence of factors other than insulin in the diabetic environment, or insulin-stimulated changes in cell types other than those studied, such as tissue macrophages or fibroblasts, which also produce insulin receptors [5]. We favour the first explanation because, in line with our results, studies in mice have shown that insulin alone stimulates effects on gene expression that are different to those produced by diabetes [31].
As predicted by our hypothesis, the most interesting finding of our study was the spatio-temporal shift in insulin response between FTs and ECs, i.e. from the maternal-facing surface at the beginning of gestation to the foetal-facing surface at term. This occurs in parallel with developmental changes in insulin receptor location [5, 32], but it is currently unclear whether this will have functional consequences at a cellular level. Thus, in addition to maternal insulin, foetal insulin can also regulate placental gene expression, and the role of foetal insulin becomes more important as gestation progresses.
The spatio-temporal changes are not only quantitative, i.e. magnitude of effect, but also qualitative: (1) the set of insulin-responsive genes was totally different between FTs and term ECs, resulting in insulin control of different processes within the same organ; and (2) the direction of gene regulation by insulin was also different between trophoblasts and ECs. Whereas in trophoblasts, genes were predominantly downregulated, in ECs, 45% of the genes were upregulated. This prevailing downregulation of expression in FTs was a striking observation; the underlying reasons are currently unknown. Genes (Table 3) that were upregulated in other studies and in FTs may serve as a positive control. This, along with the rigorous quality and viability control of the cells, strongly suggests that the observation is not due to poor cell viablility.
The differential insulin response between the maternal and foetal surface is not restricted to gene expression, but can also be observed with insulin stimulation of glycogen synthesis. At term, insulin does not lead to elevated glycogen levels in the trophoblast [33], but can stimulate glycogen synthesis in the endothelium (Desoye G, unpublished). All this underlines the cell type and tissue-specific action of insulin, even within one organ. The molecular events underlying these changes in the general response pattern are unknown and under investigation in our laboratory.
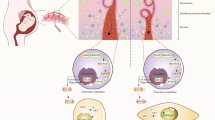
Our results indicate a shift in control of insulin-dependent processes in the placenta throughout pregnancy (Fig. 5). At the beginning of gestation, maternal insulin controls these processes. As gestation advances, the foetus gradually takes over control and will be mostly independent of maternal insulin at term. Therefore, the placenta represents a model in which insulin effects are separated in time and space via differential insulin receptor expression. This compartmentalisation of insulin effects within one organ represents, to the best of our knowledge, a novel mechanism for the control of whole-tissue function by insulin.

Spatio-temporal change of insulin effects on placental gene expression allows a shift in the control of insulin regulation from mother to foetus. In the first trimester, maternal insulin influences the placenta by interaction with trophoblast insulin receptors. These may, in turn, affect the mother by secretion of cytokines, hormones or metabolic waste products. Later in gestation, the foetus takes over control of insulin-dependent placental processes through foetal insulin interacting with placental ECs. The effects on placental development and metabolism induced at the beginning of pregnancy by maternal insulin, along with the effects of foetal insulin on the placenta later in gestation, may have repercussions on foetal development and metabolism
Abbreviations
- EC:
-
endothelial cell
- FT:
-
first-trimester trophoblast
- GDM:
-
gestational diabetes
- hCG:
-
human chorionic gonadotropin hormone
- TT:
-
term trophoblast
References
Silverman BL, Rizzo TA, Cho NH, Metzger BE (1998) Long-term effects of the intrauterine environment. The Northwestern University Diabetes in Pregnancy Center. Diabetes Care 21:B142-B149
Catalano P, Drago N (1995) Factors affecting fetal growth and body composition. Am J Obstet Gynecol 172:1459–1463
Aalinkeel R, Srinivasan M, Song F, Patel M (2001) Programming into adulthood of islet adaptations induced by early nutritional intervention in the rat. Am J Physiol Endocrinol Metab 281:E640–E648
Boloker J, Gertz S, Simmons RA (2002) Gestational diabetes leads to the development of diabetes in adulthood in the rat. Diabetes 51:1499–1506
Desoye G, Hartmann M, Blaschitz A et al (1994) Insulin receptors in syncytiotrophoblast and fetal endothelium of human placenta. Immunohistochemical evidence for developmental changes in distribution pattern. Histochemistry 101:277–285
Desoye G, Shafrir E (1996) The human placenta in diabetic pregnancy. Diabetes Rev 4:70–89
Desoye G, Myatt L (2004) The placenta. In: Reece EA, Gabbe S, Coustan D (eds) Diabetes in Women, 3rd rev edn. Lippincott, Williams & Wilkins, Baltimore, pp 147–157
Freinkel N (1980) Banting lecture 1980. Of pregnancy and progeny. Diabetes 29:1023–1035
O'Brien RM, Granner DK (1991) Regulation of gene expression by insulin. Biochem J 278:609–619
O'Brien RM, Granner DK (1996) Regulation of gene expression by insulin. Physiol Rev 76:1109–1161
O'Brien RM, Streeper RS, Ayala JE, Stadelmaier BT, Hornbuckle LA (2001) Insulin-regulated gene expression. Biochem Soc Trans 29:552–558
Cervar M, Blaschitz A, Dohr G, Desoye G (1999) Paracrine regulation of distinct trophoblast functions in vitro by placental macrophages. Cell Tissue Res 295:297–305
Lang I, Pabst MA, Hiden U et al (2003) Heterogeneity of microvascular endothelial cells isolated from human term placenta and macrovascular umbilical vein endothelial cells. Eur J Cell Biol 82:163–173
Blaschitz A, Weiss U, Dohr G, Desoye G (2000) Antibody reaction patterns in first trimester placenta: implications for trophoblast isolation and purity screening. Placenta 21:733–741
Polliotti BM, Abramowsky C, Schwartz et al (1995) Culture of first-trimester and full-term human chorionic villus explants: role of human chorionic gonadotropin and human placental lactogen as a viability index. Early Pregnancy 1:270–280
Brazma A, Hingamp P, Quackenbush J et al (2001) Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat Genet 29:365–371
Kendziorski C, Irizarry RA, Chen KS, Haag JD, Gould MN (2005) On the utility of pooling biological samples in microarray experiments. Proc Natl Acad Sci USA 22:4252–4257
Kosaki A, Pillay TS, Xu L, Webster NJ (1995) The B isoform of the insulin receptor signals more efficiently than the A isoform in HepG2 cells. J Biol Chem 270:20816–20823
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Homko C, Sivan E, Chen X, Reece EA, Boden G (2001) Insulin secretion during and after pregnancy in patients with gestational diabetes mellitus. J Clin Endocrinol Metab 86:568–573
Lindsay RS, Walker JD, Halsall I et al (2003) Scottish Multicentre Study of Diabetes in Pregnancy Insulin and insulin propeptides at birth in offspring of diabetic mothers. J Clin Endocrinol Metab 88:1664–1671
Desoye G, Hofmann HH, Weiss PA (1992) Insulin binding to trophoblast plasma membranes and placental glycogen content in well-controlled gestational diabetic women treated with diet or insulin, in well-controlled overt diabetic patients and in healthy control subjects. Diabetologia 35:45–55
Rome S, Clement K, Rabasa-Lhoret R et al (2003) Microarray profiling of human skeletal muscle reveals that insulin regulates approximately 800 genes during a hyperinsulinemic clamp. J Biol Chem 278:18063–18068
Dupont J, Dunn SE, Barrett JC, LeRoith D (2003) Microarray analysis and identification of novel molecules involved in insulin-like growth factor-1 receptor signalling and gene expression. Recent Prog Horm Res 58:325–342
Mulligan C, Rochford J, Denyer G et al (2002) Microarray analysis of insulin and insulin-like growth factor-1 (IGF-1) receptor signalling reveals the selective up-regulation of the mitogen heparin-binding EGF-like growth factor by IGF-1. J Biol Chem 277:42480–42487
Pandini G, Medico E, Conte E, Sciacca L, Vigneri R, Belfiore A (2003) Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J Biol Chem 278:42178–42189
Melis R, Pruett PB, Wang Y, Longo N (2003) Gene expression in human cells with mutant insulin receptors. Biochem Biophys Res Commun 307:1013–1020
Radaelli T, Varastehpour A, Catalano P, Hauguel-de Mouzon S (2003) Gestational diabetes induces placental genes for chronic stress and inflammatory pathways. Diabetes 52:2951–2958
Desoye G, Shafrir E (1994) Placental metabolism and its regulation in health and diabetes. Mol Aspects Med 15:505–682
Yechoor VK, Patti ME, Ueki K et al (2004) Distinct pathways of insulin-regulated versus diabetes-regulated gene expression: an in vivo analysis in MIRKO mice. Proc Natl Acad Sci USA 101:16525–16530
Jones CJ, Hartmann M, Blaschitz A, Desoye G (1993) Ultrastructural localization of insulin receptors in human placenta. Am J Reprod Immunol 30:136–145
Schmon B, Hartmann M, Jones CJ, Desoye G (1991) Insulin and glucose do not affect the glycogen content in isolated and cultured trophoblast cells of human term placenta. J Clin Endocrinol Metab 73:888–893
Acknowledgements
The authors gratefully acknowledge the excellent work of N. Prutsch, B. Hirschmugl, R. Michlmaier and S. Molzer, and would like to thank U. Lang for carefully reading the manuscript. This work was supported by the Jubilee Fund, Austrian National Bank (OeNB), Vienna (grant numbers 7,620, 10,053 and 10,896) and by the Austrian Science Foundation, Vienna (grant number 13,321)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Rights and permissions
About this article
Cite this article
Hiden, U., Maier, A., Bilban, M. et al. Insulin control of placental gene expression shifts from mother to foetus over the course of pregnancy. Diabetologia 49, 123–131 (2006). https://doi.org/10.1007/s00125-005-0054-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0054-x