
Abstract
The importance of both insulin resistance and beta cell dysfunction in the pathogenesis of glucose intolerance is widely recognised. Also popular is the concept that beta cell secretory function must be viewed in the context of extant insulin resistance. This For Debate moves from the premise that, whilst insulin action in vivo can be measured directly by a variety of essentially coherent techniques, measurement of beta cell function is more problematic. We therefore concisely survey the principal in vivo techniques that explore the diverse aspects of beta cell function and conclude that: (i) inter-correlation of clinical tests is only modest in non-diabetic subjects and poor in diabetic individuals; (ii) no single clinical test allows beta cell function to be assessed with accuracy and specificity comparable to those of insulin sensitivity; and (iii) short of complex experiments, mathematical modelling is necessary to interpret insulin secretory responses. Next we discuss the hyperbola paradigm used to describe the reciprocal relation of beta cell function to insulin sensitivity and suggest that: (i) insulin responses reflecting the basal beta cell tone are indeed inversely related to insulin action across degrees of glucose intolerance; (ii) modes of beta cell function that selectively reflect the dynamic response to acutely changing glucose concentrations are largely independent of insulin action; and (iii) when measured by experiment or resolved by modelling, quantitatively the most important of these dynamic secretion parameters is the glucose dose-response curve (glucose sensitivity). In fact, glucose excursions following glucose ingestion (i.e. glucose tolerance) are best explained by dynamic parameters of beta cell function.
Similar content being viewed by others


Introduction
The pancreatic beta cell is possibly the most complex of endocrine cells. Its main product, insulin, must be supplied to the body tissues in amounts and time-dynamics apt to maintain plasma glucose within a very narrow concentration range. To perform this task, the beta cell software features an array of specialised functions, which are finely integrated to respond to highly variable environmental inputs. In fact, insulin output must cope with size, composition and appearance rate of meals on the one hand and with target tissue sensitivity on the other. Insulin secretion rate must adapt to stimuli both on a minute-by-minute basis, like catecholamines, and in the longer term, like thyroid hormones. Thus, not only is beta cell response differentiated in relation to the stimulus, but it is also critically time-dependent. In comparison, insulin action, if equally complex at the cellular level, is a relatively stable function in any given individual. In fact, when measured in vivo by direct techniques (the euglycaemic insulin clamp), insulin sensitivity has been shown to fluctuate by 30 to 80% during 24 hours of free living [1]. With physiological or pharmacological intervention, in most cases insulin sensitivity can be at most doubled [2], whilst insulin secretion can vary many fold in the same person both in a matter of minutes (e.g. as occurs with a large mixed meal), and over years (as happens with weight gain).
One consequence of the different physiology of insulin action versus insulin secretion is that the methodology for their in vivo study is fairly advanced for the former, while it is probably less developed for the latter. In fact, in vivo insulin sensitivity can be measured by the euglycaemic clamp technique [3, 4], the accepted gold standard, or by several other methods [5], each of which basically estimates insulin-mediated glucose disappearance from the plasma under specified conditions of stimulus (=insulin concentration) and substrate (=glucose concentration). Thanks to this technological plethora, insulin sensitivity has been measured in large numbers of individuals, under disparate experimental conditions, in many disease states and in response to different pharmacological manipulations. The distribution of the trait has even been described at the population level [6, 7, 8]. Thus, we know the quantitative influence on insulin sensitivity of sex, age, body fat mass and distribution, menstrual cycle, fitness and acute physical exercise, fasting and overfeeding, diet composition and stress—to name only physiological correlates (e.g. [9, 10]). In addition, a large number of conditions have been associated with the presence of insulin resistance, Type 2 diabetes and obesity being insulin-resistant states par excellence. In comparison, our information on in-vivo insulin secretion appears to be more limited. Simply from a practical point of view, we can rank subjects by their degree of insulin sensitivity using a single number, be it a direct measure or a proxy: the position of a given subject in the rank will be reasonably certain, with a limited margin of error or fluctuation on re-testing. A similar ordering by efficiency of beta cell function would require performing multiple tests in each subject, with a definite risk that different tests would sort different ranking orders and that positions would be scrambled on repeat testing [2].
There is thus a need to re-assess the value and significance of the tests currently used to measure beta cell function in vivo and to re-analyse the physiological and pathophysiological inferences that have been based on these tests. The purpose of this article is not to repeat what can be found in several excellent reviews published in recent years [11, 12], but rather to critically examine the conceptual framework within which in vivo beta cell function is currently viewed. In particular, after briefly reviewing the main in vivo tests of insulin secretion and their inter-correlation, we shall focus on the relationship between aspects of beta cell function and insulin action in vivo. To support our arguments with numerical examples, we shall draw from the San Antonio Metabolism Study (SAM), which has recently been reported [13, 14] and analysed (E. Ferrannini, A. Gastaldelli, Y. Miyazaki et al., unpublished results), to which the reader is referred for a detailed description of the study population and primary data.
In vivo tests of insulin secretion
A preliminary issue relates to the use of peripheral plasma insulin concentrations as indicators of insulin secretion. The vast majority of the published data pertaining to beta cell function are expressed as plasma insulin concentrations [15]. Beside the problems posed by the variability and specificity (versus proinsulin and its split products) of insulin assays [16], circulating insulin concentrations are dependent upon beta cell insulin release through insulin distribution and clearance [17]. In general, the total-body volume of distribution of the hormone does not seem to vary with the insulin secretion rate, whereas whole-body insulin clearance (mostly by hepatic and renal degradation) can be regulated by the insulin concentration itself (i.e. it is a saturable process) and by insulin sensitivity of glucose uptake. Thus, insulin resistant subjects usually show a decrease in insulin clearance [18]. The application of decovolution analysis (initially used with insulin [19]) to plasma C-peptide concentrations [20] and the validation of a standard plasma C-peptide disappearance function [21] have made it possible to reconstruct insulin secretory rates from peripheral C-peptide concentrations independently of insulin clearance. Clearly, while plasma insulin concentrations are the most important determinant of insulin-mediated glucose disposal, to assess beta cell function true insulin secretion rates should be measured whenever possible.
I. Fasting plasma insulin
Fasting plasma insulin concentrations are a poor index of beta cell function for two reasons. Firstly, fasting plasma insulin does not accurately reflect pancreatic secretion, as discussed above. In fact, the relationship between the two parameters differs significantly across degrees of glucose intolerance (Fig. 1) due to a progressive decline in insulin clearance as glucose tolerance worsens. As a result, fasting insulin overestimates fasting insulin secretion in glucose intolerant compared with tolerant subjects. Secondly, fasting insulin concentrations do not take fasting glucose concentrations into account. When plotted against decreasing glucose tolerance, fasting insulin secretion shows a biphasic behaviour, rising through quartiles of NGT and IGT, peaking in mild diabetes and then gradually falling off as hyperglycaemia worsens (Fig. 2). Thus, measuring fasting insulin secretory rates is of little value in assessing overall beta cell function.

Relationship between fasting plasma insulin concentrations and fasting insulin secretory rates (ISR) in subjects with normal glucose tolerance (blue symbols), impaired glucose tolerance (green symbols) or overt Type 2 diabetes (red symbols). Each of the three slopes is significantly different from zero, but the slopes are significantly (p<0.05) different from one another (data from the SAM study)

Plot of fasting insulin levels (a) and fasting rates of insulin release (as reconstructed from C-peptide deconvolution) (b) in NGT subjects (divided into quartiles of fasting plasma glucose concentrations), in IGT subjects and in patients with Type 2 diabetes, also grouped by quartile of fasting hyperglycaemia (data from the SAM study)
Fasting serum proinsulin concentration (or its ratio to true insulin as measured by a specific assay) rises progressively across degrees of glucose intolerance [22]. This measurement is commonly regarded as a marker of inappropriate intracellular processing of pro-hormone to insulin and, by extension, as a generic index of beta cell dysfunction. Interestingly, fasting proinsulin is already markedly elevated in subjects with normal glucose tolerance who develop diabetes at follow-up [23].
II. Intravenous glucose
Intravenous glucose is generally regarded as a more specific stimulus than oral glucose to test glucose-induced insulin release. The use of intravenous glucose, however, poses problems of quantification as well as interpretation.
Intravenous glucose tolerance test
When given as a bolus (as in the IVGTT), intravenous glucose triggers a variable multiphasic secretory response, in which one or two secondary peaks, in phase with corresponding plasma glucose peaks, can often be discerned [19]. For this reason, only the first burst of insulin release is usually considered. Quantification of such first-phase insulin response (also termed ‘acute’ insulin response or AIR) has varied from the sum of insulin concentration values at multiple times (3, 5, 7, 9 and 10 min) after glucose injection [24] to the increments above baseline of plasma insulin values at 2 to 10 min [25]. Not many studies have reconstructed insulin secretion rates during an IVGTT.
Hyperglycaemic glucose clamp
When intravenous glucose is used to create a square wave of hyperglycaemia, as in the hyperglycaemic clamp technique, a biphasic insulin response is usually very clearly detectable, with a first phase, which is extinguished within ~10 min, followed by a second phase of progressively rising insulin release. Unlike the IVGTT, in which the size of the glucose bolus is well standardised, the hyperglycaemic clamp has been used to achieve varying glycaemic plateaus (from 7.2 [26] to 16.7 mmol/l [27]) and for variable periods of time, thereby eliciting very different biphasic patterns. Moreover, when comparing responses between subjects with different fasting plasma glucose concentrations, the dilemma is whether one should apply the same increment in plasma glucose or create the same hyperglycaemic plateau. The square-wave format of intravenous glucose administration usually explores only one level of steady-state hyperglycaemia. When two or more glycaemic steps are applied in succession, there is a progressive loss of AIR (Fig. 3), whereas second-phase insulin secretion is unaffected [26]. With proper standardisation, and with the proviso that the problem of comparing subjects with different fasting glucose levels remains, the hyperglycaemic clamp provides valuable indices of both first- and second-phase insulin secretion; it is, however, a complex procedure that is not widely applicable.

Plasma glucose concentrations and insulin secretion rates (ISR) during three successive steps of hyperglycaemia induced by intravenous glucose (hyperglycaemic clamp technique). Note the progressive disappearance of first-phase insulin response, with a preserved, large response to intravenous arginine. Redrawn from [26]
Graded glucose infusions
Graded intravenous glucose infusions have been used to create dose-response relationships between plasma glucose concentration and insulin secretion rate. This method has been most successfully applied by Polonsky et al. to investigate beta cell function in a variety of disease states [28]. Variations on this theme have included ramp-like increments and decrements in glucose infusion rates [29]. Although first-phase insulin secretion is not assessed, the test is a straightforward estimate of the beta cell dose-response function. Like the hyperglycaemic clamp, however, the test is not widely applicable.
Arginine test
Porte et al. [30] have pioneered the use of intravenous arginine boluses (5 g) superimposed on progressively higher, short hyperglycaemic plateaus (fasting, ~14 mmol/l and >25 mmol/l). This protocol allows one to calculate the slope of acute plasma insulin responses versus plasma glucose concentrations, termed glucose potentiation slope. This slope, expressing the sensitivity of arginine-induced acute insulin release to pre-existing glucose concentrations, is conceptually different from the slope one calculates from graded glucose infusions, which investigate mostly second-phase, or slow, insulin responses. From Porte’s protocol one can also derive a maximal (or near-maximal) response (AIRmax), which is a mixed estimate of sensitivity and responsiveness of beta cell function.
Comments
The most informative intravenous tests of beta cell function (hyperglycaemic clamp, graded glucose infusions) are quite complex and their application is necessarily limited. On the other hand, the more popular IVGTT basically tests only one specific aspect of insulin secretion, i.e. first-phase insulin release. Clearly, it is not always easy to reconcile measurements obtained with these different modes of beta cell stimulation with intravenous glucose. In addition, there are interpretative problems. Thus, years of painstaking research have not yet clarified the cellular mechanism underlying the biphasic insulin secretory response to acute glucose stimulation or its relation to beta cell dysfunction. Single-cell electrophysiological studies have shown that acute exposure to high glucose increases membrane electrical activity (extending the duration of the bursts of action potentials and simultaneously reducing the length of the silent phase) in a biphasic pattern; this enhanced electrical activity is simultaneous with spikes in cytosolic calcium concentrations and parallels insulin release both in terms of glucose-dependency and time-course [31]. Thus, it is firmly established that the classic biphasic insulin response is the compound electrophysiological result of excitatory and inhibitory (=calcium itself?) signals linked with intracellular calcium movements. However, the cellular origin of such signals is still uncertain. In addition to membrane events, intracellular processes (metabolism and second messenger generation) participate in glucose sensing and response. Amplification of the beta cell response, which generates ‘second-phase’ insulin release, is heavily dependent on intracellular steps distal to membrane ionic fluxes [31]. More importantly, calcium transients and insulin release can be partly independent of one another or even completely dissociated under certain circumstances [32]. Due to this complexity, modelling of biphasic beta cell behaviour is not well established and varies from compartmental structures with threshold distribution of glucose sensitivity [33] to more complex representations including intracellular ATP changes and mitochondrial calcium metabolism [34].
Calcium accumulation is necessary to promote exocytosis (by fusing mature secretory granules with the plasma membrane). However, morphometric studies have shown that the beta cell harbours different populations of secretory granules, in different stages of maturation and in variable spatial array between the trans-Golgi and the plasma membrane. These populations of granules constitute a chain of secretory pools in dynamic exchange with one another [35, 36]. This adds another level of complexity to beta cell function, which makes it arduous to provide a detailed description of the phenomenon and hence a plausible interpretation at the physiological level. It is known that whilst only a few of the readily releasable granules are necessary to account for the amount of insulin released in the AIR, the AIR itself is not fixed, i.e. it increases with progressively larger boluses and can be further augmented by other hormones (e.g. GLP-1 [37]). In other words, the whole endocrine pancreas behaves like a single beta cell, i.e. with a short-lived secretory burst, again suggesting simultaneous triggering of an inhibitory stimulus responsible for the rapidly descending arm of the peak [38]. It is not clear what happens to AIR—with the discharge of readily releasable granules—when the glucose stimulus is more gradual: i.e. whether the phenomenon is simply slowed down or a different phenomenon is called into action (or a combination of the two).
Interpretation of the second phase of in vivo insulin response to intravenous glucose is more straightforward, but its quantification requires maintaining plasma glucose levels at the pre-set plateau (as with the hyperglycaemic clamp) for a pre-set period of time. Dose-response functions created from multiple hyperglycaemic steps or graded infusions do not formally account for glucose memory: antecedent glycaemic exposure can modify the secretory response to the same glucose level.
III. Oral glucose
Administration of glucose (or a mixed meal) by mouth is generally reputed to be a less specific test of beta cell function than intravenous glucose, despite its obvious physiological superiority. This is because other secretagogues (both nutrients and gut hormones) concur with glucose to stimulate the beta cell; in addition, the levels of the stimuli change continually with time. Whether and how the seemingly constitutive biphasic beta cell response to acute glucose stimulation contributes to the time-course of insulin secretion in response to oral glucose cannot be resolved. A popular index of acute insulin response to oral glucose is the insulinogenic index, or the insulin : glucose ratio at some early time following glucose (or mixed meal) ingestion. Variations on this theme abound: ratios of absolute concentrations or their increments above baseline, at 30 min or at 15 min or some other period of time into the OGTT can all be found in the literature. Other indices derived from an OGTT curve are the ratio of insulin : glucose area under the curve (or the respective increments above baseline). These empirical indices all reflect the need to adjust insulin secretion for the concomitant glucose concentration.
When oral glucose is superimposed on a hyperglycaemic plateau established with intravenous glucose, the step-up in insulin secretion rate is a measure of the incretin effect (Fig. 4). Alternatively, one may simulate the plasma glucose response to oral glucose by a variable intravenous glucose infusion and then take the difference in the respective insulin responses as a measure of the incretin effect.

A square wave of hyperglycaemia (+4 mmol/l) elicits a biphasic insulin secretory response. Ingestion of a glucose load while maintaining the glycaemic plateau causes a detectable further rise in insulin secretion. Redrawn from [57]
Comments
The insulin secretory pattern following oral glucose is the integrated response to a more complex set of factors than are called upon by intravenous glucose. Furthermore, the stimulus (glucose, other nutrients, secretagogues) is continually changing. Therefore, some form of mathematical modelling must be applied to formally account for the changing glycaemia and dissect the different modes of response of the beta cell.
Inter-correlation of in vivo tests of insulin secretion
Correlation between different in vivo tests of insulin secretion is generally unsatisfactory. Thus, in a group of 16 subjects with variable degrees of glucose intolerance, measurements for beta cell function derived from the OGTT or a continuous intravenous glucose infusion correlated only moderately (r values of 0.5–0.7) with AIR and second-phase insulin secretion as calculated from a hyperglycaemic clamp [39]. More recently, Stumvoll et al. [25] reported an extended correlation matrix of various indices of insulin secretion derived from fasting measurements, a hyperglycaemic clamp and an OGTT, showing correlation coefficients ranging from 0.22 to 0.79 in a group of 104 non-diabetic subjects. While previous studies have mostly examined the correlation between various indices in non-diabetic subjects, the largest discrepancy is found in diabetic patients. For example, in a group of patients with Type 2 diabetes, in whom both an OGTT and an IVGTT were carried out a few days apart from each other, inter-correlation of the fasting insulin secretion rates measured on the occasion of the two studies was excellent. In contrast, there was no correlation at all between the amount of insulin secreted during the first 30 min after glucose ingestion and the AIR on the IVGTT (Fig. 5). These data suggest that the IVGTT may not be the ideal test to assess beta cell function in diabetes. Possibly, in a compromised beta cell the strong inhibitory signal generated by an abrupt and large rise in plasma glucose overcomes stimulatory signals and masks a secretory response that a slower stimulus, like the OGTT, would otherwise reveal. As previously mentioned, intracellular calcium itself may be the ‘toxic’ agent at play under the circumstances of an IVGTT.

a Correlation between basal insulin secretion rates (ISR) measured in patients with Type 2 diabetes on different days (r=0.87, p<0.0001). b Lack of correlation between insulin secretion between 0–30 min on the OGTT and the acute insulin response (AIR) on the IVGTT in the same patients (r=0.17, p=NS). For both the OGTT and IVGTT, insulin secretion rates were calculated from C-peptide measurements by deconvolution
More generally, one wonders whether the AIR, probably the most common empirical index of insulin secretion, which has generated precious information (i.e. the progression to diabetes in IGT individuals [40]), may have overestimated the degree of beta cell incompetence in diabetes and, consequently, have underestimated the extent to which beta cell function could be improved in these patients. Clearly, these issues remain to be tested experimentally.
Having recognised the complexity of beta cell function and the empirical nature of the available clinical tests, Porte et al. have advocated the application of a panel of tests to rank an individual’s beta cell status and follow it up through intervention [41]. This is a logical predicament, but also one that can be applied only in the settings of clinical research.
Mathematical modelling
Since the early studies on beta cell function in the isolated perfused rat pancreas and in humans, mathematical models have been developed to describe insulin secretion [33, 42, 43, 44]. These models, particularly those by Licko and Grodsky [33, 42] and Cerasi et al. [44], have established the fundamental mathematical constructs necessary to describe beta cell function. However, these sophisticated models were not devised as tools to quantify beta cell function in an individual. The first attempt at a model-based description of beta cell function test of wider applicability was made by Toffolo et al. [45] using IVGTT data. This model enriched the characterisation of beta cell function by adding a parameter of second-phase insulin secretion to the classic first-phase response, obviously within the limitations inherent in the IVGTT. More recent modelling approaches have exploited the OGTT or a meal test, and rely on C-peptide rather than insulin concentrations to calculate insulin secretion [29, 46, 47, 48, 49]. These models follow the paradigm illustrated in Figure 6. Insulin secretion is represented as the sum of two principal components: one expressing the fact that insulin secretion is a function of glucose concentration (the dose-response function), and one accounting for the observation that rapid changes in glucose concentration enhance insulin release (represented as a function of the glucose concentration derivative). In one class of models (Fig. 6a), it is postulated that glucose concentration changes are reflected in insulin secretion with some delay [29, 46, 47]. Another line of thinking [48, 49] (Fig. 6b) allows for modulation of the dose-response due to phenomena known as potentiation [48, 50]. The beta cell model is coupled to a model of C-peptide kinetics (typically that of [21]), so that the whole model predicts C-peptide concentration as a function of glucose concentration. Beta cell function parameters, such as slope and intercept of the dose-response function, can thus be determined from the analysis of C-peptide and glucose concentrations measured during a variety of clinical tests.

Mathematical models of beta cell function (see text for explanation)
Although these models share some common features, an important difference concerns the representation of potentiation of insulin secretion, which is substantial during an oral test. Time-dependent potentiation of insulin secretion is a phenomenon that is apparent even with intravenous glucose [51]: when glucose concentration is raised to a fixed hyperglycaemic level, insulin secretion exhibits a progressive increase with time in spite of constant glucose (Fig. 4). When glucose is administered orally, not only is time-dependent potentiation present, but insulin secretion is also potentiated by the gastrointestinal hormones (mainly GIP and GLP-1) (Fig. 4). Potentiation, which has been described mathematically by Cerasi et al. for intravenous glucose [44, 52], is included only in model b of Figure 6. Model a depicts the sustained insulin secretion at the end of an oral test (or intravenous test, [29]) as a first-order delay (i.e. a single linear differential equation). Although this may be a practical assumption, it does not seem to correspond to a well-defined beta cell physiological event [51]. Furthermore, experiments in the perfused pancreas [43] clearly show that a square wave of hyperglycaemia produces a slow, progressive stimulation but a quick shut-off of insulin secretion; this behaviour is not compatible with a first-order delay model, in which speed of onset and offset are the same. Whether the different models of Figure 6 yield convergent interpretations of the same OGTT data remains, however, to be established. Beyond the unavoidable limitations of individual models, the modelling approach does underscore the fact that beta cell function cannot be reduced to a single parameter (as typically done with the empirical indices): beta cell responses comprise diverse components, which may be interdependent but are not necessarily an expression of the same cellular phenomenon. For example, the model of [48, 49] (cf. Fig. 6b) yields parameters of beta cell function that quantify the sensitivity of the insulin response to the initial rapid glucose rise (rate sensitivity), the slope of the dose-response (glucose sensitivity), and the magnitude of potentiation, expressed as the fold increment of this factor. As illustrated in the following section, a key finding of the analysis of beta cell function with this model is that the model indices are differentially related to insulin sensitivity and glucose tolerance.
Relationship between insulin secretion and insulin resistance
The hyperbola paradigm
The relationship between insulin resistance and beta cell function is of great interest because of the physiological feedback linking the two functions in vivo. The obvious example of the operation of such a feedback is obesity: in non-diabetic subjects, both basal ISR and total insulin release during an OGTT increase linearly with BMI (Fig. 7). The current interpretation of this relationship is that the insulin resistance of obese individuals signals back to the beta cell to enhance its insulin output in order to maintain normal glucose tolerance. Thus, a logical evolution of this notion has been to formally relate insulin secretion to insulin action. Kahn et al. [53] have pioneered the concept that a hyperbolic relationship exists between beta cell function and insulin sensitivity, and that constancy of their product across a wide range of beta cell responses and insulin sensitivity is key to maintaining glucose tolerance. Using data obtained from an IVGTT-minimal model in young, healthy subjects, the plots of the insulin sensitivity index SI against (i) fasting plasma insulin, (ii) AIR (as the insulin increments during the first 10 min after intravenous glucose), (iii) the potentiation slope or (iv) AIRmax (derived from Porte’s protocol [30]) could all be adequately fit with power functions (of the general form β=a·σb, where β and σ stand for beta cell function and insulin sensitivity respectively). Because the exponents (b in the equation) of these fitting functions were very close to −1 (ranging from −0.84 to −1.19), the authors concluded that in general the relationship between insulin sensitivity and beta cell function is best described by a hyperbola (β=a·σ−1=a/σ) (Fig. 8), i.e. a non-linear function such that insulin release changes relatively little over a wide span of insulin sensitivity but then rises dramatically with further decrements in insulin sensitivity. The interpretation was that glucose tolerance is preserved as long as the beta cell can compensate for decreased insulin action by appropriately increasing insulin release. Because a characteristic of the hyperbolic equation β=a·σ−1 is the constancy of the product β·σ, which equals the coefficient ‘a’, a disposition index (DI=β·σ=a, i.e. the product of insulin sensitivity and insulin secretory response) was recommended as an inclusive tool of quantifying glucose disposition in vivo. The hyperbola paradigm has been applied in several studies (cf. [11]), including longitudinal analyses of glucose tolerance in Pima Indians [54]. The predictive power of DI for the development of diabetes in population studies has not been tested, however.

Linear increase in basal (fasting) insulin secretion rate (circles, in pmol·min−1·m−2) and total insulin output (squares, in nmol·m−2) during a 3-hour OGTT in non-diabetic subjects grouped by quartile of BMI (unpublished data)

Theoretical hyperbolic relationship (β=a·σ−1) between insulin secretion (β) and insulin sensitivity (σ). The function is linearised by a log–log transformation (log[β]=log[a]−log[σ]). As hyperbolas differ only in intercept, each point (=subject) can be described by a single parameter, called disposition index (DI=a=σ·β). DI decreases (= curves regress towards the axes origin) as glucose tolerance worsens
The hyperbola approach presents some problems. Firstly, whether the fitting function is a hyperbola or other depends not only on the number and quality of experimental data but also on the nature of the variables, i.e. the way in which insulin sensitivity and insulin secretion are expressed. If the hyperbola β=a·σ−1 holds true for the beta cell function and insulin sensitivity indices β and σ, just the use of a different insulin sensitivity index σ′, linearly related but not proportional to σ (e.g. σ=σ′+1), results in a different relationship (β=a·(σ′+1)−1). The DI paradigm is not valid in the latter relationship, as β·σ′ is no longer constant. This point is better illustrated with the use of the data in Table 1, where a large dataset—including euglycaemic insulin clamp measurements of insulin-mediated glucose uptake and the insulin response to an OGTT—is analysed using different expressions of insulin sensitivity (the straight M value from the clamp or insulin-mediated glucose clearance corrected for the steady-state plasma insulin concentration, MCRG/I) and three different empirical indices of beta cell function (fasting insulin, total plasma insulin response to the OGTT [AUCI] and the insulinogenic index [ΔI30/ΔG30]). Whereas a non-linear fit is statistically superior to a linear fit and in most cases the best fit is a power function, the exponent can be significantly different from −1 (ranging from −0.7 to −1.8, with r values between 0.46 and 0.75) even just in subjects with NGT depending on how insulin response and insulin sensitivity are expressed. Secondly, when examining individuals with IGT or diabetes (Fig. 9), not only do the coefficients of the fitting power functions differ significantly from those of NGT subjects, but so too do the exponents (Table 1): if hyperbolas were the best fit for each group, only the coefficient would differentiate IGT and diabetes from NGT (in other words, the curves would move towards the axis origin, cf. Fig. 8).

Power functions (with 95% CI) describing the relationship between the insulinogenic index (from the OGTT) and the M value (from a euglycaemic insulin clamp) in NGT (blue lines), IGT (green lines) and diabetic subjects (red lines) (parameters given in Table 1)
This mathematical consideration has the following corollaries. Comparing DIs of different groups of subjects may be less sensitive than comparing both coefficients and exponents of the respective power functions (as done in Table 1). Furthermore, Stumvoll et al. [55] have argued that even in subjects with NGT, ‘compensation’ may be incomplete: using data obtained in Pima Indians with normal glucose tolerance, they reported that moving leftward along the same hyperbola both fasting and 2-h plasma glucose concentrations increase (albeit minimally) despite a constant DI [55]. In other words, in insulin-resistant subjects in whom AIR is increased enough to keep the product DI constant glucose tolerance is slightly worse (an average increase in fasting glucose of 0.2 mmol/l) than in insulin-sensitive subjects with the same DI. These authors have used the term glucose allostasis for this phenomenon, to indicate that the feedback of insulin action on insulin secretion must involve a change in the chronic level of the signal (i.e. the plasma glucose concentration), and have proposed that the ratio of AIR : M be calculated as an index of beta cell demand (BCDI, or the compensatory load imposed on the beta cell). Note that BCDI (AIR/M) and DI (AIR·M) are generated from the same variables.
The hyperbola paradigm has proven extraordinarily expedient in directing attention to the fact that there are modes of beta cell response that are coupled to insulin action in the intact organism. It also allows one to estimate quickly (via the DI) the compensatory increase in some indices of insulin secretion in response to insulin resistance. For example, for the three groups of subjects plotted in Figure 9 the median DIs (as M·Δ30I/Δ30G, from Table 1) are 4.8, 3.1 and 0.28 mmol·min−1·kgFFM −1·[pmol/mmol] in NGT, IGT and diabetic subjects respectively (p<0.0001): this compensation, whatever the underlying mechanism, is progressively worse as glucose tolerance declines. The situation, however, may be more complex.
Functions of the beta cell and their role in glucose tolerance
To illustrate the complexity of the beta cell response, we first examine the relationship between insulin sensitivity and indices of tonic beta cell function. When insulin sensitivity is plotted against basal (post-absorptive) insulin secretion rate, one power function (β=307·σ−0.38, r=0.43, p<0.0001) best describes the relationship in NGT, IGT and diabetic subjects alike (Table 2, Fig. 10a). Thus, compensation for insulin resistance of the basal secretory tone occurs adequately regardless of glucose tolerance (even in markedly hyperglycaemic patients). Clearly, diabetic subjects have higher fasting plasma glucose concentrations; if their basal insulin secretion could be assessed at normoglycaemia as in the two other groups, diabetic subjects would likely fall below the compensation line. Thus, from the mechanistic viewpoint the ‘compensation’ of basal ISR that is seen in diabetic subjects is only apparent.

Plot of basal insulin secretion rate (ISR) (a) and total insulin output during an OGTT (b) against insulin sensitivity in NGT (blue symbols), IGT (green symbols) and diabetic subjects (red symbols)
If we then analyse the relation of insulin resistance to total insulin output during the OGTT, diabetic subjects fall on a very different power function as compared to NGT or IGT subjects (Fig. 10b, Table 2). Thus, a higher basal insulin secretory activity is generally associated with a higher total stimulated insulin output, as is predicted to happen in a secretory system whose set point is raised. The relationship, however, breaks down as fasting hyperglycaemia ensues (Fig. 11), presumably a reflection of a blockade of insulin processing (cf. elevated proinsulin levels) and release and/or a defect in the maximal secretory capacity (≈AIRmax in Porte’s terminology).

Relationship of total insulin output during an OGTT to basal insulin secretion rate in NGT subjects (blue symbols, divided into quartiles of mean OGTT plasma glucose concentrations), in IGT subjects (green symbol) and in patients with Type 2 diabetes, also grouped by quartile of post-glucose hyperglycaemia (red symbols)
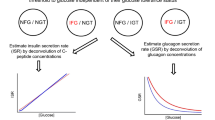
If we now analyse the relationship between insulin resistance and a dynamic index of beta cell function, namely the mean slope of the glucose-ISR dose-response curve (or glucose sensitivity, as calculated by model-based analysis of OGTT data, cf. previous section), we see a completely different picture. Glucose sensitivity declines drastically from NGT to IGT to diabetes (from median values of 101 to 55 to 13 pmol·min·m−2·mmol/l−1) but bears little relation to insulin resistance (Fig. 12a and Table 2). The same holds true for rate sensitivity, i.e. the anticipation of insulin release by speed of change in plasma glucose concentrations. Thus, the ability of the beta cell to respond to meal-induced glucose increments is not dependent on—or, at least, not closely related to—insulin action. This result is in keeping with the finding reported by Polonsky et al. using their graded glucose infusions protocol, that the dose-response curves in obese (hence insulin-resistant) and lean non-diabetic women [56] are almost parallel; this implies that in insulin resistance the set-point for insulin response to intravenous glucose is shifted upward but without any major changes in glucose sensitivity.

Plot of glucose sensitivity (i.e. slope of the glucose-ISR dose-response curve) against insulin resistance (a) or mean plasma glucose during the OGTT (b) in NGT (blue symbols), IGT (green symbols) and diabetic subjects (red symbols). While unrelated to insulin resistance (p=NS), glucose sensitivity is strongly associated with mean OGTT glycaemia (y=3.5·104·x -3.0, r=0.81, p<0.0001)
From the analysis of these sample data—and with the proviso that validation in different data-sets is necessary— indices of tonic beta cell activity (best exemplified by the basal insulin secretion rate) are possibly linked to insulin sensitivity by feedback mechanism(s), for which plasma glucose itself, plasma NEFA and gastro-intestinal hormones are candidate signals. On the other hand, indices of dynamic beta cell response to acute stimulation, such as glucose sensitivity and rate sensitivity, are less dependent on insulin action as they appear to reflect membrane and post-membrane phenomena triggered by glucose stimulation at any set point of the secretory machinery. The discrepancy between the results illustrated above (Fig. 12a) and those obtained with AIR [53] or the insulinogenic index (Fig. 9) depends on the fact that the beta cell does not respond to insulin resistance by up-regulating all its functions. Clearly, the release of insulin after an intravenous glucose bolus (AIR) is mediated by some but not all the cellular mechanisms that mediate insulin release during a meal. On the other hand, the insulinogenic index is a composite marker of both early release, possibly related to AIR, and dynamic characteristics of beta cell function (dose-response increments in release). As such, it is expected to be variably related to insulin sensitivity (cf. Table 1).
The crucial question then arises: what is the relation of the various indices of beta cell function to glucose tolerance? An approximate answer can be provided by examining the statistical contribution of each of them to glucose tolerance, i.e. the extent to which they explain the variability of glucose tolerance. By using pooled data from the sample data in NGT, IGT and diabetic subjects, we can calculate that glucose sensitivity alone explains more than 50% of the variability in mean glucose levels during the OGTT (see power function in Fig. 12b). More precisely, when analysing glucose concentrations at different times during the OGTT by a multiple regression including several variables (Table 3), glucose sensitivity is always tightly related to glucose levels; this is also the case for the other two dynamic indices (rate sensitivity and potentiation). Basal and total insulin release are only weakly associated with glycaemia, and the pattern of independent associations is essentially unaltered by including insulin sensitivity in the statistical model (Table 3).
In summary, while the closest correlate of insulin sensitivity is the basal secretory tone, glucose sensitivity, which is largely unrelated to insulin sensitivity, is a stronger predictor of glucose tolerance than the basal secretory tone. This means that beta cell function cannot be reduced to a single index, that the different modes of beta cell function affect glucose tolerance differently.
Conclusions
Whereas different in vivo tests of insulin sensitivity essentially estimate the same phenomenon (insulin-driven flux of glucose into cells), different in vivo tests of insulin secretion explore very different aspects of beta cell function. Because inter-correlation of the insulin secretion indices is modest or, in the case of diabetic individuals, poor, no single in vivo test allows beta cell function to be assessed with accuracy and specificity comparable to those of insulin sensitivity.
Mathematical modelling is necessary to interpret insulin secretory responses in terms of relevant parameters of beta cell function. Models can be used to analyse intravenous glucose tests, but secretory responses to intravenous glucose may be paradoxical in subjects with diabetes. Use of OGTT (or mixed meal) data may be preferable not only for simplicity but also for physiological significance.
The hyperbola paradigm has been useful to bring into focus the relationship between insulin secretion and insulin resistance. In general, power functions can quantify the feedback of insulin resistance on beta cell responses, particularly those reflecting the beta cell tone or set point. There are, however, modes of beta cell function that seem to be largely independent of insulin action. These are secretion parameters that reflect the dynamic response to changing glucose levels over a time frame of minutes or hours; the most important of these is the glucose dose-response curve.
Glucose excursions after glucose ingestion (i.e. glucose tolerance) are best explained by dynamic parameters of beta cell function. Linear (or non-linear) combinations of such parameters and insulin sensitivity may explain very large fractions of the variability of glucose curves.
More work, both theoretical and experimental, is needed before the critical analysis we present here can be consolidated and accepted. Mathematical models can be refined and need further validation. Experiments must include accounts of different nutrients and secretagogues. The stakes, however, are high. A better understanding of the complexities of in vivo behaviour of the endocrine pancreas is already an ambitious goal. Even more challenging is the prospect of providing answers to two most important issues in diabetes, namely, how much beta cell mass versus beta cell function is lost in diabetic subjects, and how much of either can be retrieved by medical intervention.
Abbreviations
- AIR:
-
acute insulin response
- DI:
-
disposition index
- GLP-1:
-
glucagon-like peptide-1
- ISR:
-
insulin secretion rate
- M:
-
whole-body insulin-mediated glucose uptake
- MCR:
-
metabolic clearance rate
References
Morgan LM, Aspostolakou F, Wright J, Gama R (1999) Diurnal variations in peripheral insulin resistance and plasma non-esterified fatty acid concentrations: a possible link? Ann Clin Biochem 36:447–450
Ferrannini E (1998) Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: problems and prospects. Endocr Rev 19:477–490
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 273:E214–E223
Ferrannini E, Mari A (1998) How to measure insulin sensitivity. J Hypertens 16:895–906
Bergman RN, Finegood DT, Ader M (1985) Assessment of insulin sensitivity in vivo. Endocr Rev 6:45–86
Ferrannini E, Vichi S, Beck-Nielsen H, Laakso M, Paolisso G, Smith U (1996) Insulin action and age. European Group for the Study of Insulin Resistance (EGIR). Diabetes 45:947–953
Haffner SM, Howard G, Mayer E et al. (1997) Insulin sensitivity and acute insulin response in African-Americans, non-Hispanic whites, and Hispanics with NIDDM: the Insulin Resistance Atherosclerosis Study. Diabetes 46:63–69
Clausen JO, Borch-Johnsen K, Ibsen H et al. (1996) Insulin sensitivity index, acute insulin response, and glucose effectiveness in a population-based sample of 380 young healthy Caucasians. J Clin Invest 98:1195–1209
Camastra S, Bonora E, Del Prato S, Rett K, Weck M, Ferrannini E (1999) Effect of obesity and insulin resistance on resting and glucose-induced thermogenesis in man. EGIR (European Group for the Study of Insulin Resistance). Int J Obes Relat Metab Disord 23:1307–1313
Howard BV, Mayer-Davis EJ, Goff D et al. (1998) Relationships between insulin resistance and lipoproteins in nondiabetic African Americans, Hispanics, and non-Hispanic whites: the Insulin Resistance Atherosclerosis Study. Metabolism 47:1174–1179
Kahn SE (2003) The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 46:3–19
McClenaghan NH, Flatt PR (1999) Physiological and pharmacological regulation of insulin release: insights offered through exploitation of insulin-secreting cell lines. Diabetes Obes Metab 1:137–150
Ferrannini E, Gastaldelli A, Miyazaki Y et al. (2003) Predominant role of reduced b-cell sensitivity to glucose over insulin resistance in impaired glucose tolerance. Diabetologia 46:1211–1219
Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, DeFronzo RA (2004) Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia 47:31–39
Kahn SE, Porte D Jr (1997) The pathophysiology of type II (noninsulin-dependent) diabetes mellitus: implications for treatment. In: Porte D Jr, Sherwin RS (eds) Ellenberg and Rifkin’s diabetes mellitus: theory and practice, 5th edn. Appleton & Lange, Stamford, pp 487–512
Robbins DC, Andersen L, Bowsher R et al. (1996) Report of the American Diabetes Association’s Task Force on standardization of the insulin assay. Diabetes 45:242–256
Ferrannini E, Cobelli C (1987) The kinetics of insulin in man. I. General aspects. Diab Metab Rev 3:335–363
Ferrannini E, Natali A, Bell P, Cavallo-Perin P, Lalic N, Mingrone G (1997) Insulin resistance and hypersecretion in obesity. J Clin Invest 100:1166–1173
Ferrannini E, Pilo A (1979) Pattern of insulin delivery after intravenous glucose injection in man and its relation to plasma glucose disappearance. J Clin Invest 64:243–254
Eaton RP, Allen RC, Schade DS, Erickson KM, Standefer J (1980) Prehepatic insulin production in man: kinetic analysis using peripheral connecting peptide behavior. J Clin Endocrinol Metab 51:520–528
Van Cauter E, Mestrez F, Sturis J, Polonsky KS (1992) Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 41:368–377
Mykkanen L, Zaccaro DJ, Hales CN, Festa A, Haffner SM (1999) The relation of proinsulin and insulin to insulin sensitivity and acute insulin response in subjects with newly diagnosed type II diabetes: the Insulin Resistance Atherosclerosis Study. Diabetologia 42:1060–1066
Haffner SM, Gonzalez C, Mykkanen L, Stern M (1997) Total immunoreactive proinsulin, immunoreactive insulin and specific insulin in relation to conversion to NIDDM: the Mexico City Diabetes Study. Diabetologia 40:830–837
Bergman RN, Phillips LS, Cobelli C (1981) Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest 68:1456–1467
Stumvoll M, Mitrakou A, Pimenta W et al. (2000) Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care 23:295–301
Toschi E, Camastra S, Sironi AM et al. (2002) Effect of acute hyperglycemia on insulin secretion in man. Diabetes 51 [Suppl 1]:S130–S133
DeFronzo RA, Ferrannini E, Hendler R, Felig P, Wahren J (1983) Regulation of splanchnic and peripheral glucose uptake by insulin and hyperglycemia in man. Diabetes 32:35–45
Polonsky KS (1995) Lilly Lecture 1994. The beta-cell in diabetes: from molecular genetics to clinical research. Diabetes 44:705–717
Toffolo G, Breda E, Cavaghan MK, Ehrmann DA, Polonsky KS, Cobelli C (2001) Quantitative indexes of beta-cell function during graded up&down glucose infusion from C-peptide minimal models. Am J Physiol 280:E2–E10
Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr (1984) Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 74:1318–1328
Mears D, Atwater I (2000) Electrophysiology of the pancreatic β-cell. In: LeRoith D, Taylor S, Olefsky J (eds) Diabetes mellitus. A fundamental and clinical text, 2nd edn. Lippincott Williams & Wilkins, Philadelphia, pp 47–61
Atwater I, Carroll P, Li M (1989) Electrophysiology of the pancreatic β-cell. In: Draznin B, Melmed S, LeRoith D (eds) Molecular and cellular biology of diabetes mellitus. Alan R. Liss, New York, p 49
Grodsky GM (1972) A threshold distribution hypothesis for packet storage of insulin and its mathematical modeling. J Clin Invest 51:2047–2059
Wollheim CB, Sharp GW (1981) Regulation of insulin release by calcium. Physiol Rev 61:914–973
Bratanova-Tochkova TK, Cheng H, Daniel S et al. (2002) Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes 51 [Suppl 1]:S83–S90
Henquin JC, Ishiyama N, Nenquin M, Ravier MA, Jonas JC (2002) Signals and pools underlying biphasic insulin secretion. Diabetes 51 [Suppl 1]:S60–S67
Vahl TP, Paty BW, Fuller BD, Prigeon RL, D’Alessio DA (2003) Effects of GLP-1-(7-36)NH2, GLP-1-(7-37), and GLP-1-(9-36)NH2 on intravenous glucose tolerance and glucose-induced insulin secretion in healthy humans. J Clin Endocrinol Metab 88:1772–1779
Jonkers FC, Henquin JC (2001) Measurements of cytoplasmic Ca2+ in islet cell clusters show that glucose rapidly recruits beta-cells and gradually increases the individual cell response. Diabetes 50:540–550
Nijpels G, Wal PS van der, Bouter LM, Heine RJ (1994) Comparison of three methods for the quantification of beta-cell function and insulin sensitivity. Diabetes Res Clin Pract 26:189–195
Warram JH, Sigal RJ, Martin BC, Krolewski AS, Soeldner JS (1996) Natural history of impaired glucose tolerance: follow-up at Joslin Clinic. Diabet Med 13:S40–S45
Porte D Jr (1996) Normal physiology and phenotypic characterization of beta-cell function in subjects at risk for non-insulin-dependent diabetes mellitus. Diabet Med 13:S25–S32
Licko V (1973) Threshold secretory mechanism: a model of derivative element in biological control. Bull Math Biol 35:51–58
Bergman RN, Urquhart J (1971) The pilot gland approach to the study of insulin secretory dynamics. Recent Prog Horm Res 27:583–605
Cerasi E, Fick G, Rudemo M (1974) A mathematical model for the glucose induced insulin release in man. Eur J Clin Invest 4:267–278
Toffolo G, Bergman RN, Finegood DT, Bowden CR, Cobelli C (1980) Quantitative estimation of beta cell sensitivity to glucose in the intact organism: a minimal model of insulin kinetics in the dog. Diabetes 29:979–990
Breda E, Cavaghan MK, Toffolo G, Polonsky KS, Cobelli C (2001) Oral glucose tolerance test minimal model indexes of beta-cell function and insulin sensitivity. Diabetes 50:150–158
Cretti A, Lehtovirta M, Bonora E et al. (2001) Assessment of beta-cell function during the oral glucose tolerance test by a minimal model of insulin secretion. Eur J Clin Invest 31:405–416
Mari A, Schmitz O, Gastaldelli A, Oestergaard T, Nyholm B, Ferrannini E (2002) Meal and oral glucose tests for the assessment of b-cell function: modeling analysis in normal subjects. Am J Physiol 283:E1159–E1166
Mari A, Tura A, Gastaldelli A, Ferrannini E (2002) Assessing insulin secretion by modeling in multiple-meal tests: role of potentiation. Diabetes 51 [Suppl 1]:S221–S226
Mari A (2002) Mathematical modeling in glucose metabolism and insulin secretion. Curr Opin Clin Nutr Metab Care 5:495–501
Nesher R, Cerasi E (1987) Biphasic insulin release as the expression of combined inhibitory and potentiating effects of glucose. Endocrinology 121:1017–1024
Cerasi E (1981) Differential actions of glucose on insulin release: re-evaluation of a mathematical model. In: Cobelli C, Bergman RN (eds) Carbohydrate metabolism. Quantitative physiology and mathematical modelling. Wiley, Chichester, pp 3–22
Kahn SE, Prigeon RL, McCulloch DK et al. (1993) Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42:1663–1672
Weyer C, Bogardus C, Mott DM, Pratley RE (1999) The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 104:787–794
Stumvoll M, Tataranni PA, Stefan N, Vozarova B, Bogardus C (2003) Glucose allostasis. Diabetes 52:903–909
Byrne M, Sturis J, Cavaghan M, O’Meara N, Polonsky K (2000) Insulin secretion in humans. Physiologic regulation and alterations in disease states. In: LeRoith D, Taylor S, Olefsky J (eds) Diabetes mellitus. A fundamental and clinical text, 2nd edn. Lippincott Williams & Wilkins, Philadelphia, pp 105–114
Natali A, Gastaldelli A, Galvan AQ et al. (1998) Effects of acute alpha 2-blockade on insulin action and secretion in humans. Am J Physiol 274:E57–E64
Acknowledgements
This work was supported by an EFSD-Novo Nordisk Type 2 Programme Focused Research Grant and funds from the Italian Ministry of University and Scientific Research (MURST prot. 2001065883_001). We thank Dr Ralph DeFronzo of the University of Texas Health Science Center at San Antonio, Texas for making available to us the San Antonio Metabolism Study database to exemplify our arguments, and for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferrannini, E., Mari, A. Beta cell function and its relation to insulin action in humans: a critical appraisal. Diabetologia 47, 943–956 (2004). https://doi.org/10.1007/s00125-004-1381-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1381-z