
Abstract
Color is one of the phenotypic markers mostly used to study soybean (Glycine max L. Merr.) genetic, molecular and biochemical processes. Two P450-dependent mono-oxygenases, flavonoid 3′-hydroxylase (F3′H; EC1.14.3.21) and flavonoid 3′,5′-hydroxylase (F3′5′H, EC1.14.13.88), both catalyzing the hydroxylation of the B-ring in flavonoids, play an important role in coloration. Previous studies showed that the T locus was a gene encoding F3′H and the W1 locus co-segregated with a gene encoding F3′5′H in soybean. These two genetic loci have identified to control seed coat, flower and pubescence colors. However, the allelic distributions of both F3′H and F3′5′H genes in soybean were unknown. In this study, three novel alleles were identified (two of four alleles for GmF3′H and one of three alleles for GmF3′5′H). A set of gene-tagged markers was developed and verified based on the sequence diversity of all seven alleles. Furthermore, the markers were used to analyze soybean accessions including 170 cultivated soybeans (G. max) from a mini core collection and 102 wild soybeans (G. soja). For both F3′H and F3′5′H, the marker selection efficiencies for pubescence color and flower color were determined. The results showed that one GmF3′H allele explained 92.2 % of the variation in tawny and two gmf3′h alleles explained 63.8 % of the variation in gray pubescence colors. In addition, two GmF3′5′H alleles and one gmF3′5′h allele explained 94.0 % of the variation in purple and 75.3 % in white flowers, respectively. By the combination of the two loci, seed coat color was determined. In total, 90.9 % of accessions possessing both the gmf3′h-b and gmf3′5′h alleles had yellow seed coats. Therefore, seed coat colors are controlled by more than two loci.
Similar content being viewed by others


Introduction
Flower, seed and fruit colors are important in the ecology of plants and in their ability to attract pollinators and seed-dispersing organisms (Grotewold 2006; Mol et al. 1998). In addition, color is one of the most widely used phenotypic markers in the study of genetic, molecular and biochemical processes, due to their easy recognizability (Holton and Cornish 1995; Koes et al. 2005). The pigments that color most flowers, fruits and seeds are of three types: betalains, carotenoids, and flavonoids (Tanaka et al. 2008). Of these, the flavonoids have been studied most in the context of pigmentation, and are well conserved among higher plants (Hichri et al. 2011; Lepiniec et al. 2006; Winkel-Shirley 2001). Flavonoids mainly consist of anthocyanins, chalcone, flavone, flavonol, flavanone and isoflavonoids. Of these, anthocyanins are broadly distributed in flowering plants and predominantly contribute to flower, fruit and seed colors (Han et al. 2010). Beside their multiple roles in coloring plant organs, flavonoids are of great interest for plant adaptation to the environment and for human health (Halbwirth 2010; Harvaux and Kloppstech 2001; Li et al. 1993; Pourcel et al. 2007; Scalbert et al. 2005; Winkel-Shirley 2002).
The biosynthetic pathway of flavonoids is well established and many of the structural and some of the regulatory genes have been cloned in several model plants including Arabidopsis, maize (Zea mays), snapdragon (Antirrhinum majus) and Petunia (Hichri et al. 2011; Holton and Cornish 1995; Mol et al. 1998; Winkel-Shirley 2001). The precursors for the synthesis of all flavonoids are malonyl-CoA and p-coumaroyl-CoA. Chalcone synthase catalyzes the stepwise condensation of three acetate units from malonyl-CoA with p-coumaroyl-CoA to yield tetrahydroxychalcone. The synthesis of colored anthocyanins from tetrahydroxychalcone is catalyzed by a series of enzymes, including chalcone isomerase, flavanone 3-hydroxylase, flavonoid 3′-hydroxylase (F3′H), flavonoid 3′,5′-hydroxylase (F3′5′H), dihydroflavonol 4-reductase, anthyocyanidin synthase and uridine diphosphate glucose: flavonoid-3-O-glucosyltransferase (Holton and Cornish 1995). Among these, F3′H and F3′5′H, which are microsomal cytochrome P450-dependent mono-oxygenases that require NADPH as a co-factor, are key enzymes to hydroxylate the B-ring in flavonoids (Forkmann 1991). As the hydroxylation pattern of the B-ring in flavonoids plays an important role in coloration and determines their stability and antioxidant capacity, the activity of F3′H and F3′5′H strongly influences flower and seed coloration (de Vetten et al. 1999; Han et al. 2010; Holton et al. 1993; Ishiguro et al. 2011; Moreau et al. 2012; Schoenbohm et al. 2000; Zabala and Vodkin 2003). To date, the F3′H and F3′5′H genes have been isolated from many plant species, including Petunia (Brugliera et al. 1999; Holton et al. 1993), Arabidopsis (Schoenbohm et al. 2000), soybean (Toda et al. 2002; Zabala and Vodkin 2007), grape (Bogs et al. 2006; Falginella et al. 2010), pea (Moreau et al. 2012), maize (Sharma et al. 2011), and apple (Han et al. 2010). Furthermore, manipulation of F3′H and F3′5′H has been effective in the genetic engineering of floral crops to develop new genotypes with novel flower colors for ornamental purposes (Nakatsuka et al. 2007; Tanaka et al. 2010).
The chemistry and genetics of pigmentation in seeds and flowers has also been extensively studied in soybean (Palmer and Stelly 1979; Stephens and Nickell 1992; Todd and Vodkin 1993), Thus far, alleles of at least five loci (I, T, W1, R, and O) are known to function epistatically to control seed coat pigmentation, six loci (W1, W2, W3, W4, Wm, and Wp) control flower pigmentation and two loci (T and Td) control pubescence color in soybean (Palmer et al. 2004; Stephens and Nickell 1992). So far, the molecular biology of these soybean loci has been studied but only a few loci have been identified at the molecular level due to the complexity of the soybean genome. Classic genetics showed that T conferred brown and t conferred gray pubescence color. Further, T generally darkens hilum and/or seed coat color in combination with genotypes at the I, W1, R and O loci (Palmer et al. 2004). Cloning and mapping of the soybean sf3′h1 genomic and cDNA sequences identified the F3′H gene at the T locus on linkage group C2 (chromosome 6) (Toda et al. 2002; Zabala and Vodkin 2003). Silencing of sf3′h1 resulted in decreased levels of quercetin and loss of the tawny pigmentation in pubescence (Nagamatsu et al. 2009; Nagamatsu et al. 2007), suggesting that the sf3′h1 gene is responsible for the production of quercetin and involved in the control of pigmentation in pubescence. Moreover, the relationship between the T locus and chill-tolerance has also been studied (Takahashi and Asanuma 1996; Takahashi et al. 2005; Toda et al. 2012), indicating the importance of the soybean F3′H gene in pigment biosynthesis and physiological function. The W1 locus has a pleiotropic effect on flower and hypocotyl color, in which soybean accessions with purple/white flowers have purple/green hypocotyls. It was confirmed that W1 locus co-segregated with a gene encoding F3′5′H by using near isogenic lines (Zabala and Vodkin 2007). In addition, the light-purple flower of the wild soybean B09121 is controlled by a new allele of the W1 locus encoding F3′5′H (Takahashi et al. 2010), suggesting that more alleles of these genes remain to be identified.
The sequences of F3′H and F3′5′H have been cloned from some soybean accessions; however, no functional markers and allelic diversity analysis of these genes have been published so far. Due to the abundant variation in the soybean genome, whether F3′H and F3′5′H have other alleles and their distribution in accessions need to be revealed. The core collection and mini core collection of soybean provide an effective platform for genetic diversity studies, novel gene identification, and allele distribution analysis (Song et al. 2010; Wang et al. 2006). In this study, three novel alleles, two of four alleles for GmF3′H and one of three alleles for GmF3′5′H, were found on the basis of the sequence variation of these two genes in different soybean accessions. A set of gene-tagged markers were developed and verified. The genetic effects of GmF3′H and GmF3′5′H were also detected by genotyping 272 accessions including part of the mini core collection of cultivated soybeans and annual wild soybeans.
Materials and methods
Plant materials
The collection of annual wild soybeans (Glycine soja) and a partial mini core collection of cultivated soybeans (Glycine max) including elite cultivars and local landraces described previously (Li et al. 2008) were obtained from the National Genebank at the Institute of Crop Science, Chinese Academy of Agricultural Sciences. Two kinds of pubescence colors and flower colors, and five kinds of seed coat colors (Supplemental Fig. 1) were classified according to Qiu and Chang (2006).
RNA extraction, cDNA cloning and sequencing
Total RNA from frozen leaves of soybean ecotype Willimas82 and Zhonghuang13 was extracted using TRIzol reagent (Invitrogen, USA). To eliminate the contamination of genomic DNA, total RNA was treated with RNase-free DNase (TaKaRa, Japan). The cDNA was synthesized using the ReverTra Ace qPCR RT kit (Toyobo, Japan) in a reaction of 20 μL. The full-coding cDNA clones of GmF3′H and GmF3′5′H were generated by RT-PCR and the primers used to amplify the coding sequences were listed in Supplemental Table 1. The coding sequences were determined after cloning into the pBluescript SK + (pBS) vector, and confirmed by comparison with the corresponding genomic sequences.
Multiple sequence alignments and phylogenetic tree building
To generate the phylogenetic tree of F3′Hs and F3′5′Hs from different organisms, the amino-acid sequences of F3′Hs and F3′5′Hs identified previously were collected from the NCBI database (http://www.ncbi.nlm.nih.gov). Additional amino-acid sequences of F3′Hs were from: Arabidopsis thaliana (AAG16746), Petunia × hybrida (AAD56282), Vitis vinifera (CAI54278), Brassica napus (ABC58723), Antirrhinum majus (ABB53383), Malus × domestica (ACR14867), Zea mays (AEF33624), Sorghum bicolor (AAV74195), and Oryza sativa (AAP52914). Additional amino-acid sequences of F3′5′Hs were: Petunia × hybrida (CAA80265), Gentiana scabra (BAE86871), Pisum sativum (ADW66160), Antirrhinum kelloggii (BAJ16328), Vitis vinifera (CAI54277), Solanum tuberosum (AAV85470), Solanum lycopersicum (ADC80513), Phalaenopsis hybrid (AAZ79451), Dendrobium moniliforme (AEB96145), and Hordeum vulgare (BAK02913). All sequences were aligned with ClustalX and improved manually. Phylogenetic trees were constructed by the neighbor-joining method using MEGA 4.0 and internal branch support was estimated with 1,000 bootstrap replicates.
Genomic DNA isolation and PCR for sequencing
Genomic DNA was isolated from soybean leaves using the DNeasy Plant Mini Kit (Qiagen, USA) and used at 10–20 ng per PCR amplification. PCR was carried out using KOD-Plus-Neo (Toyobo, Japan) according to the manufacturer’s recommendations in a PTC-200 Thermocycler (MJ Research/Bio-Rad, USA). The PCR reaction cycles were as follows: 1 cycle (94 °C, 3 min), 36 cycles (94 °C, 15 s; 60 °C, 15 s; 68 °C, 30 s), and a final extension step (68 °C, 8 min). PCR products were analyzed by gel electrophoresis to verify the size and ensure specific amplification, and then sequenced after isolation. The primers used for amplification and sequencing are listed in Supplementary Table 1.
Marker development
Gene-tagged markers were developed based on the sequence variation of different alleles. CAPs markers F3′H-ApoI and F3′5′H-HphI were developed based on single-nucleotide deletion of adenine at position 973 of the GmF3′H coding sequence and single-nucleotide substitution at position 1424 of the GmF3′5′H coding sequence. The dCAP markers F3′H-MjaIV and F3′H-EcoNI were developed based on the artificial introduction of a restriction enzyme-recognition site at the end of the forward primer. The InDel marker F3′5′H-In was developed based on the 53-bp difference between the two alleles of GmF3′5′H.
Genotype analysis
PCR was carried out in the PTC-200 Thermocycler using the genomic DNA of all materials. PCR products were directly separated on 2 % agarose gels for the InDel marker or digested with the appropriate restriction enzymes for the CAPS and dCAPS markers. The samples were incubated at the temperature recommended by the manufacturer for >1 h, and then separated on 2 % agarose gels stained with ethidium bromide followed by photography.
Results
Cloning of the GmF3′H and GmF3′5′H genes from soybean and phylogenetic analysis
The full-length protein-coding sequences for GmF3′H (Glyma06g21920) from soybean ecotype Williams 82 and GmF3′5′H (Glyma13g04210) from Zhonghuang13 were obtained by RT-PCR. Sequence comparison of the cloned coding sequences and the published genomic sequence indicated the presence of three exons and two introns in these genes. Sequence analysis revealed a putative polypeptide consisting of 513 amino-acids from the coding sequence of GmF3′H, and one of 509 amino-acids from GmF3′5′H.
To study the relationship of GmF3′H and GmF3′5′H with other F3′H/F3′5′H proteins, phylogenetic analysis was carried out using the deduced amino-acid sequences of GmF3′H and GmF3′5′H with other known plant-specific flavonoid hydroxylase proteins from Arabidopsis, Petunia, grape, rapeseed (Brassica napus), snapdragon, apple, maize, grain sorghum (Sorghum bicolor), rice (Oryza sativa), and so on. The phylogenetic tree was separated into two large groups, with all F3′Hs and F3′5′Hs clustered in the different clades (Fig. 1). GmF3′H/GmF3′5′H resided in the same clade as other dicots, and were separated from the monocots such as maize, grain sorghum, rice, P. hybrid and D. moniliforme (Fig. 1). Moreover, F3′H/F3′5′H from the most closely related species formed closely related clades, such as Arabidopsis and rapeseed, maize and grain sorghum.

Phylogenetic tree of F3′Hs and F3′5′Hs proteins from soybean and other species. Neighbor-joining tree of F3′Hs and F3′5′H proteins from soybean and other species generated using MEGA 4.0. The numbers on each node are bootstrap values, which indicate the percentage of bootstrap replicates that support this node out of 1,000 samples. Branch lengths are proportional to the estimated number of amino-acid substitutions. Scale bar indicates the estimated amino-acid substitutions per site. F3′H superfamily; Arabidopsis thaliana (AAG16746), Petunia x hybrida (AAD56282), Vitis vinifera (CAI54278), Brassica napus (ABC58723), Antirrhinum majus (ABB53383), Malus × domestica (ACR14867), Zea mays (AEF33624), Sorghum bicolor (AAV74195), and Oryza sativa (AAP52914). F3′5′H superfamily; Petunia × hybrida (CAA80265), Gentiana scabra (BAE86871), Pisum sativum (ADW66160), Antirrhinum kelloggii (BAJ16328), Vitis vinifera (CAI54277), Solanum tuberosum (AAV85470), Solanum lycopersicum (ADC80513), Phalaenopsis hybrid (AAZ79451), Dendrobium moniliforme (AEB96145), Hordeum vulgare (BAK02913)
Allelic diversity in the GmF3′H and GmF3′5′H genes
To study the sequence diversity of the coding regions in the GmF3′H and GmF3′5′H genes, 30 soybean accessions (12 modern elite cultivars, ten local landraces and eight annual wild soybeans) were used for sequence analysis, and four alleles of GmF3′H and three alleles of GmF3′5′H were found (Table 1; Figs. 2, 3).

GmF3′H gene structure and polymorphisms in different alleles. The gmf3′h-a1 allele presents a single base deletion of adenine at coding sequence position 973 relative to the start codon. The deletion creates a premature stop codon and a truncated protein of 394 residues compared to the reference GmF3′H protein of 513 residues. The gmf3′h-a2 allele presents a single base insertion of an adenine at position 965 as well as a single base deletion of adenine at position 973, resulting in N322 K and Q324T changes. The gmf3′h-b allele presents a single base deletion of a cytosine at position 1164. This cytosine deletion resulted in a frame-shift that prematurely truncated the protein of 394 residues. Boxes represent exons; lines between boxes represent introns; boxes with dashed lines represent exons with untranslated regions in predicted proteins from different alleles

GmF3′5′H gene structure and polymorphisms in different alleles. The GmF3′5′H-b allele had three nucleotide substitutions in exon 3 (C1059A, T1424A, C1509T): two synonymous and one non-synonymous substitution (V475E). The gmf3′5′h allele had a 53-bp fragment insertion at coding sequence position 1352 relative to the start codon, resulting in a prematurely truncated amino-acid sequence of 467 residues compared to the reference GmF3′5′H protein of 501 residues. Boxes represent exons; lines between boxes represent introns; boxes with dashed lines represent exons with untranslated regions in predicted proteins from different alleles
In the gmf3′h-a1 allele from Zhongpin03-5373, Zhonghuang13 and Jidou-12, a single base deletion of an adenine at coding sequence position 973 relative to the start codon resulted in a premature stop codon and a truncated protein of 394 residues (Fig. 2). In the gmf3′h-a2 allele from Shang951099 and Yingdehedou, a single base insertion of an adenine at coding sequence position 965 as well as a single base deletion of an adenine at position 973 only resulted in N322K and Q324T changes for no frame-shift with the single base deletion right after the single base insertion. In the gmf3′h-b allele from Wenfeng7, Suinong14, Suinong20, Zheng92116 and Pixiansilicao, a single base deletion of a cytosine was identified at position 1164. This deletion resulted in a frame-shift that prematurely truncated the protein after only six amino-acids. The GmF3′H alleles in 20 other accessions (designated GmF3′H) were the same as the reference Williams 82 sequence and encoded the full-length amino-acids.
In the gmf3′5′h allele in Williams 82, Wenfeng7, Union, JiNF58, Suinong20, Baiqiu1 and Peking, a 53-bp fragment was inserted at coding sequence position 1352 relative to the start codon, resulting in a prematurely truncated amino-acid sequence of 467 residues (Fig. 3). In the other cultivars/lines, the GmF3′5′H alleles from 14 accessions (designated GmF3′5′H-a) were the same as the sequence of Zhonghuang13 and encoded the full-length amino-acids while those in another nine accessions (GmF3′5′H-b) had three nucleotide substitutions in exon 3 (C1059A, T1424A, C1509T) resulting in two synonymous and one non-synonymous substitution (V475E).
Development of gene-tagged markers
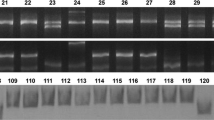
Molecular markers were designed to distinguish the alleles of GmF3′H and GmF3′5′H (Table 2). For the gmf3′h-a1 and gmf3′h-a2 alleles, a CAPS marker designated as F3′H-ApoI was generated based on the single nucleotide deletion of adenine at coding sequence position 973. At the deletion site, the GmF3′H and gmf3′h-b alleles could be recognized by ApoI, whereas the gmf3′h-a1 and gmf3′h-a2 alleles could not be recognized by this enzyme. The PCR results showed that the products were 540 bp in length as expected, and the PCR products of the GmF3′H and gmf3′h-b alleles were digested into two fragments (322 and 208 bp) by ApoI, whereas the products of the gmf3′h-a1 and gmf3′h-a2 alleles were not cleaved. Hence, the F3′H-ApoI marker could be used to identify both the gmf3′h-a1 and gmf3′h-a2 alleles (Fig. 4a). To distinguish the gmf3′h-a1 and gmf3′h-a2 alleles, a dCAPs marker was developed, since no restriction enzyme recognition sites were detected at the insertion site of adenine at position 965. A restriction enzyme recognition site (GTNNAC, MjaIV) was artificially introduced at the end of the forward primer, containing two mismatched nucleotides (GT; Table 2). The PCR results showed that the expected DNA fragments (133/132 bp) were easily amplified using materials with gmf3′h-a1 and gmf3′h-a2 alleles. The PCR products were then digested by the MjaIV enzyme. The product of the gmf3′h-a1 allele was digested, and a 105-bp fragment was obtained. On the other hand, the PCR product of the gmf3′h-a2 allele could not be cleaved by MjaIV (Fig. 4b). We concluded that the F3′H-MjaIV marker can be used to separate the two alleles. For the gmf3′h-b allele, a dCAPS marker was also developed due to the absence of restriction enzyme recognition sites at the deletion site. Similarly, the recognition site of EcoNI (CCTNNNNNAGG) was artificially introduced at the end of the forward primer, containing only one mismatched nucleotide (C; Table 2). The PCR products were amplified and then digested by the EcoNI enzyme. The product of the gmf3′h-b allele could not be digested and gave a 167-bp fragment, while that of the other alleles was digested and a 140-bp fragment was obtained (Fig. 4c).

Polymorphisms revealed by three gene-tagged markers of GmF3′H. The F3′H-ApoI marker distinguished gmf3′h-a1 and non-gmf3′h-a1 alleles (a); the F3′H-MjaIV marker distinguished gmf3′h-a2 and non-gmf3′h-a2 alleles (b); the F3′H-EcoNI marker distinguished gmf3′h-b and non-gmf3′h-b alleles (c)
For the gmf3′5′h allele, an InDel marker was developed based on the 53-bp difference between gmf3′5′h and GmF3′5′H. The PCR results revealed that the GmF3′5′H-In marker successfully differentiated the gmf3′5′h and GmF3′5′H alleles by the amplification of 395- and 342-bp DNA fragments, respectively, as predicted (Fig. 5a). To distinguish the GmF3′5′H-a and GmF3′5′H-b alleles, a CAPS marker, F3′5′H-HphI, was developed based on a single-nucleotide substitution (T to A) at coding sequence position 1424. At the substitution site, the GmF3′5′H-a allele could be recognized by HphI whereas the GmF3′5′H-b allele could not. The PCR products were amplified and then digested by HphI. The product of the GmF3′5′H-b allele could not be digested and gave a 342-bp fragment, while that of the GmF3′5′H-a allele was digested and 225- and 117-bp fragments were obtained (Fig. 5b).

Polymorphisms revealed by two gene-tagged markers of GmF3′5′H. The F3′5′H-In marker distinguished gmf3′5′h and non-gmf3′5′h alleles (a); the F3′5′H-HphI marker distinguished GmF3′5′H-a and GmF3′5′H-b alleles (b)
The developed markers F3′H-ApoI, F3′H-MjaIV, F3′H-EcoNI, F3′5′H-In and F3′5′H-HphI were verified by analysis of allelic diversity using the 30 soybean accessions. The results suggested that the identification of molecular markers was consistent with the sequencing data. Further, these markers can be easily used on a large scale in genotyping basically dependent on the PCR technique.
Distribution of alleles of GmF3′H and GmF3′5′H in soybean subgenus soja
To determine the genetic constitution of the GmF3′H and GmF3′5′H alleles in the soybean subgenus soja, the developed markers were used to genotype 272 accessions: 170 cultivated soybeans (G. max) from mini core collection, consisting of 24 modern elite cultivars and 146 local landraces, and 102 wild soybeans (G. soja) (Table 3, Supplemental File). All the wild soybeans contained the GmF3′H allele. The ranking of GmF3′H allele frequencies in cultivars and landraces was similar, with GmF3′H > gmf3′h-b > gmf3′h-a1 > gmf3′h-a2. GmF3′H was the most common allele in cultivars (45.8 %) and landraces (63.0 %) and gmf3′h-b (29.2 and 28.1 %, respectively) was the second main allele. With regard to the GmF3′5′H locus, the most common allele in wild soybeans was GmF3′5′H-a (92.2 %). Only six wild soybeans possessed GmF3′5′H-b and two had gmf3′5′h. However, the ranking of GmF3′5′H allele frequencies in cultivars and landraces differed from wild soybeans, with gmf3′5′h > GmF3′5′H-b > GmF3′5′H-a. Nei’s gene diversity index analysis suggested that the diversity of both GmF3′H and GmF3′5′H was higher in cultivated than in wild soybeans (Table 3).
Genetic effects of GmF3′H and GmF3′5′H genes
Previous studies showed that the T locus was a gene encoding F3′H and the W1 locus co-segregated with a gene encoding F3′5′H in soybean, which belonged to the genetic loci controlling seed coat, flower and pubescence color. The genetic effect of GmF3′H alleles on pubescence color and seed coat was estimated (Table 4). The results showed that >80 % of accessions possessing the GmF3′H (86.3 %) or gmf3′h-a2 (83.3 %) alleles had a tawny pubescence color while >80 % of those possessing the gmf3′h-a1 (84.6 %) or gmf3′h-b (83.3 %) alleles had a gray pubescence color. As to seed coat color, ~45.4 % accessions possessing GmF3′H had a black seed coat, which was the most widespread seed coat color of the GmF3′H allele. However, most accessions possessing gmf3′h-a1, gmf3′h-a2 or gmf3′h-b had a yellow seed coat while few had other seed coat colors. On the other hand, these results also suggested that one GmF3′H allele explained 177/192 (92.2 %) of tawny and two gmf3′h alleles (gmf3′h-a1 and gmf3′h-b) explained 51/80 (63.8 %) of gray colors.
For the F3′5′H locus, 92.3 % of accessions possessing GmF3′5′H-a and 88.9 % possessing GmF3′5′H-b had a purple flower while 82.1 % of those possessing gmf3′5′h had a white flower (Table 5). With regard to seed coat color, ~54.9 % accessions possessing GmF3′5′H-a had a black seed coat, which was also the most widespread seed coat color of the GmF3′5′H allele. However, the most widespread seed coat color of the gmf3′5′h allele was yellow (73.1 %). The relationship between GmF3′5′H-b and seed coat color was not very close and the frequency of each color was similar. Two GmF3′5′H alleles and one gmF3′5′h allele explained 187/199 (94.0 %) of purple and 55/73 (75.3 %) of white flowers.
When the genetic effect of the GmF3′H and GmF3′5′H alleles were combined, the results showed that these loci had a high association with seed coat color (Table 6). Sixty-five percent of accessions possessing both GmF3′H and GmF3′5′H-a had a black seed-coat and 90.9 % of those possessing both gmf3′h-b and gmf3′5′h had a yellow seed-coat, contributions higher than the single locus.
Discussion
Previous studies showed that GmF3′H and GmF3′5′H had at least two alleles, one (designated GmF3′H and GmF3′5′H-a in this study) encoding the whole amino-acid sequences and the other (designated gmf3′h-b and gmf3′5′h in this study) encoding prematurely terminated sequences due to a frame-shift (Zabala and Vodkin 2003; Zabala and Vodkin 2007). These two kinds of alleles of GmF3′H/GmF3′5′H were associated with the T/W1 loci, respectively (Toda et al. 2002; Zabala and Vodkin 2007). In our research, four alleles including two novel alleles of GmF3′H and three alleles including one novel allele of GmF3′5′H were identified in soybean accessions. Among these, one allele of GmF3′H (gmf3′h-a1) also encoded a premature termination due to a single base deletion of adenine at coding sequence position 973 relative to the start codon (Fig. 2). The site of the base deletion in gmf3′h-a1 was in front of that in the allele of GmF3′H identified previously, resulting in an earlier occurrence of the frame-shift. The other novel alleles identified in this study (gmf3′h-a2 and GmF3′5′H-b) resulted from non-synonymous nucleotide substitutions. Association analysis of allele and phenotype showed that these two alleles had no influence on the corresponding gene.
Although the genetic diversity of G. soja was reduced 50 % by the bottleneck of domestication (Hyten et al. 2006), it appears that selection for GmF3′H and GmF3′5′H did not cause erosion of diversity. This was inferred by the finding of four GmF3′H alleles among cultivated soybeans, whereas G. soja only contained the GmF3′H allele. With regard to GmF3′5′H, nearly all wild soybeans (92.2 %) contained the GmF3′5′H-a allele while three GmF3′5′H alleles occurred among cultivated soybeans. The distribution of these alleles in cultivated and wild soybean was similar to an artificial selection locus GmTfl1, which is associated with growth habit (Tian et al. 2010). These results suggested that the GmF3′H and GmF3′5′H loci might also have undergone artificial selection along with wild soybeans to landraces and then cultivars. However, this kind of selection might go with the selection of seed coat color since these two genes are all associated with seed coat color.
Functional marker development is very important for gene discovery in crops. A polymorphism study of the IFS1 and IFS2 genes indicated that three SNPs in IFS1 and two SNPs in IFS2 were closely associated with all individual types and total seed isoflavone concentrations (Cheng et al. 2008). In the present study, gene-tagged markers of GmF3′H and GmF3′5′H were developed and verified. Analysis of the genotype in 272 soybeans including the partial mini core collection showed that GmF3′H was correlated with pubescence color and GmF3′5′H was correlated with flower color. Therefore, these gene-tagged markers can be used to genotype the cultivars on the target locus. By PCR and/or digestion, the alleles of GmF3′H and GmF3′5′H could be clearly separated, indicating that nearly all SNPs or InDels could be used for developing CAPS/dCAPs markers. However, the phenotypes of some accessions were still not correlated with the genotypes, differing from previous reports (Toda et al. 2002; Zabala and Vodkin 2007). This was due to the existence of other loci controlling pubescence, flower and seed coat color in soybean and/or the existence of other alleles of these two genes. For example, another locus Td is also related to the control of pubescence color in soybean, and some of the cultivars may have the Td/td allele that is near grey as exemplified in this study. All wild soybeans possessed the GmF3′H allele, indicating that the Td locus may not have effect in the G. soja accessions. In addition, the dominant I allele exhibits a completely colorless seed coat phenotype due to dominance inhibition possibly via a posttranscriptional mode of gene silencing (Senda et al. 2004). The T and R loci determine specific seed coat color only in combination with the recessive i allele. Therefore, this interaction also reduced the frequency of GmF3′H alleles in the surveyed accessions. Using partial mini core collection, three new alleles, gmf3′h-a1, gmf3′h-a2 and GmF3′5′H-b, were identified, and their frequencies were 4.4, 2.6, and 23.2 %, which indicated that the materials we used for genotyping had abundant genetic variation.
References
Bogs J, Ebadi A, McDavid D, Robinson SP (2006) Identification of the flavonoid hydroxylases from grapevine and their regulation during fruit development. Plant Physiol 140:279–291
Brugliera F, Barri-Rewell G, Holton TA, Mason JG (1999) Isolation and characterization of a flavonoid 3′-hydroxylase cDNA clone corresponding to the Ht1 locus of Petunia hybrida. Plant J 19:441–451
Cheng H, Yu O, Yu D-Y (2008) Polymorphisms of IFS1 and IFS2 gene are associated with isoflavone concentrations in soybean seeds. Plant Sci 175:505–512
de Vetten N, ter Horst J, van Schaik HP, de Boer A, Mol J, Koes R (1999) A cytochrome b5 is required for full activity of flavonoid 3′, 5′-hydroxylase, a cytochrome P450 involved in the formation of blue flower colors. Proc Natl Acad Sci USA 96:778–783
Falginella L, Castellarin SD, Testolin R, Gambetta GA, Morgante M, Di Gaspero G (2010) Expansion and subfunctionalisation of flavonoid 3′,5′-hydroxylases in the grapevine lineage. BMC Genomics 11:562–579
Forkmann G (1991) Flavonoids as flower pigments: the formation of the natural spectrum and its extension by genetic engineering. Plant Breed 106:1–26
Grotewold E (2006) The genetics and biochemistry of floral pigments. Annu Rev Plant Biol 57:761–780
Halbwirth H (2010) The creation and physiological relevance of divergent hydroxylation patterns in the flavonoid pathway. Int J Mol Sci 11:595–621
Han Y, Vimolmangkang S, Soria-Guerra RE, Rosales-Mendoza S, Zheng D, Lygin AV, Korban SS (2010) Ectopic expression of apple F3′H genes contributes to anthocyanin accumulation in the Arabidopsis tt7 mutant grown under nitrogen stress. Plant Physiol 153:806–820
Harvaux M, Kloppstech K (2001) The protective functions of carotenoid and flavonoid pigments against excess visible radiation at chilling temperature investigated in Arabidopsis npq and tt mutants. Planta 213:953–966
Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V (2011) Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot 62:2465–2483
Holton TA, Cornish EC (1995) Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell 7:1071–1083
Holton TA, Brugliera F, Lester DR, Tanaka Y, Hyland CD, Menting JG, Lu CY, Farcy E, Stevenson TW, Cornish EC (1993) Cloning and expression of cytochrome P450 genes controlling flower colour. Nature 366:276–279
Hyten DL, Song Q, Zhu Y, Choi IY, Nelson RL, Costa JM, Specht JE, Shoemaker RC, Cregan PB (2006) Impacts of genetic bottlenecks on soybean genome diversity. Proc Natl Acad Sci USA 103:16666–16671
Ishiguro K, Taniguchi M, Tanaka Y (2011) Functional analysis of Antirrhinum kelloggii flavonoid 3′-hydroxylase and flavonoid 3′,5′-hydroxylase genes; critical role in flower color and evolution in the genus Antirrhinum. J Plant Res 125:451–456
Koes R, Verweij W, Quattrocchio F (2005) Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci 10:236–242
Lepiniec L, Debeaujon I, Routaboul JM, Baudry A, Pourcel L, Nesi N, Caboche M (2006) Genetics and biochemistry of seed flavonoids. Annu Rev Plant Biol 57:405–430
Li J, Ou-Lee TM, Raba R, Amundson RG, Last RL (1993) Arabidopsis flavonoid mutants are hypersensitive to UV-B irradiation. Plant Cell 5:171–179
Li Y, Guan R, Liu Z, Ma Y, Wang L, Li L, Lin F, Luan W, Chen P, Yan Z, Guan Y, Zhu L, Ning X, Smulders MJ, Li W, Piao R, Cui Y, Yu Z, Guan M, Chang R, Hou A, Shi A, Zhang B, Zhu S, Qiu L (2008) Genetic structure and diversity of cultivated soybean (Glycine max (L.) Merr.) landraces in China. Theor Appl Genet 117:857–871
Mol J, Grotewold E, Koes R (1998) How genes paint flowers and seeds. Trends Plant Sci 3:212–217
Moreau C, Ambrose MJ, Turner L, Hill L, Ellis TH, Hofer JM (2012) The b gene of pea encodes a defective flavonoid 3′,5′-hydroxylase, and confers pink flower color. Plant Physiol 159:759–768
Nagamatsu A, Masuta C, Senda M, Matsuura H, Kasai A, Hong JS, Kitamura K, Abe J, Kanazawa A (2007) Functional analysis of soybean genes involved in favonoid biosynthesis by virus induced gene silencing. Plant Biotechnol 5:778–790
Nagamatsu A, Masuta C, Matsuura H, Kitamura K, Abe J, Kanazawa A (2009) Down-regulation of flavonoid 3′-hydroxylase gene expression by virus-induced gene silencing in soybean reveals the presence of a threshold mRNA level associated with pigmentation in pubescence. J Plant Physiol 166:32–39
Nakatsuka T, Abe Y, Kakizaki Y, Yamamura S, Nishihara M (2007) Production of red-flowered plants by genetic engineering of multiple flavonoid biosynthetic genes. Plant Cell Rep 26:1951–1959
Palmer RG, Stelly DM (1979) Reference diagrams of seed coat colors and patterns for use as genetic markers in crosses. Soybean Genet Newsl 6:55–57
Palmer RG, Pfeiffer TW, Buss GR, Kilen TC (2004) Soybeans: improvement, production, and uses, 3rd edn. ASA, CSSA, and SSSA, Madison (WI)
Pourcel L, Routaboul JM, Cheynier V, Lepiniec L, Debeaujon I (2007) Flavonoid oxidation in plants: from biochemical properties to physiological functions. Trends Plant Sci 12:29–36
Qiu L-J, Chang R-Z (2006) Descriptors and data standard for soybean (Glycine spp.), China Agriculture Press, Beijing
Scalbert A, Johnson IT, Saltmarsh M (2005) Polyphenols: antioxidants and beyond. Am J Clin Nutr 81:215S–217S
Schoenbohm C, Martens S, Eder C, Forkmann G, Weisshaar B (2000) Identification of the Arabidopsis thaliana flavonoid 3′-hydroxylase gene and functional expression of the encoded P450 enzyme. Biol Chem 381:749–753
Senda M, Masuta C, Ohnishi S, Goto K, Kasai A, Sano T, Hong JS, MacFarlane S (2004) Patterning of virus-infected Glycine max seed coat is associated with suppression of endogenous silencing of chalcone synthase genes. Plant Cell 16:807–818
Sharma M, Cortes-Cruz M, Ahern KR, McMullen M, Brutnell TP, Chopra S (2011) Identification of the pr1 gene product completes the anthocyanin biosynthesis pathway of maize. Genetics 188:69–79
Song X-E, Li Y-H, Chang R-Z, Guo P-Y, Qiu L-J (2010) Population structure and genetic diversity of mini core collection of cultivated soybean (Glycine max (L.) Merr.) in China. Sci Agricul Sin 43:2209–2219
Stephens PA, Nickell CD (1992) Inheritance of pink flower in soybean. Crop Sci 32:1131–1132
Takahashi R, Asanuma S (1996) Association of T gene with chilling tolerance in soybean. Crop Sci 36:559–562
Takahashi R, Benitez ER, Funatsuki H, Ohnishi S (2005) Soybean maturity and pubescence color genes improve chilling tolerance. Crop Sci 45:1387–1393
Takahashi R, Dubouzet JG, Matsumura H, Yasuda K, Iwashina T (2010) A new allele of flower color gene W1 encoding flavonoid 3′5′-hydroxylase is responsible for light purple flowers in wild soybean Glycine soja. BMC Plant Biol 10:155–164
Tanaka Y, Sasaki N, Ohmiya A (2008) Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J 54:733–749
Tanaka Y, Brugliera F, Kalc G, Senior M, Dyson B, Nakamura N, Katsumoto Y, Chandler S (2010) Flower color modification by engineering of the flavonoid biosynthetic pathway: practical perspectives. Biosci Biotechnol Biochem 74:1760–1769
Tian Z, Wang X, Lee R, Li Y, Specht JE, Nelson RL, McClean PE, Qiu L, Ma J (2010) Artificial selection for determinate growth habit in soybean. Proc Natl Acad Sci USA 107:8563–8568
Toda K, Yang D, Yamanaka N, Watanabe S, Harada K, Takahashi R (2002) A single-base deletion in soybean flavonoid 3′-hydroxylase gene is associated with gray pubescence color. Plant Mol Biol 50:187–196
Toda K, Kuroiwa H, Senthil K, Shimada N, Aoki T, Ayabe SI, Shimada S, Sakuta M, Miyazaki Y, Takahashi R (2012) The soybean F3′H protein is localized to the tonoplast in the seed coat hilum. Planta 236:79–89
Todd JJ, Vodkin LO (1993) Pigmented soybean (Glycine max) seed coats accumulate proanthocyanidins during development. Plant Physiol 102:663–670
Wang L, Guan Y, Guan R, Li Y, Ma Y, Dong Z, Liu X, Zhang H, Zhang Y, Liu Z, Chang R, Xu H, Li L, Lin F, Luan W, Yan Z, Ning X, Zhu L, Cui Y, Piao R, Liu Y, Chen P, Qiu L (2006) Establishment of Chinese soybean (Glycine max) core collections with agronomic traits and SSR markers. Euphytica 151:215–223
Winkel-Shirley B (2001) Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol 126:485–493
Winkel-Shirley B (2002) Biosynthesis of flavonoids and effects of stress. Curr Opin Plant Biol 5:218–223
Zabala G, Vodkin L (2003) Cloning of the pleiotropic T locus in soybean and two recessive alleles that differentially affect structure and expression of the encoded flavonoid 3′ hydroxylase. Genetics 163:295–309
Zabala G, Vodkin LO (2007) A rearrangement resulting in small tandem repeats in the F3′5′H gene of white flower genotypes is associated with the soybean W1 locus. Crop Sci 47:S113–S124
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31071446 and 31271753) and the State Key Basic Research and Development Plan of China (973) (2010CB125900). The authors thank Dr. Iain C Bruce (University of Hong Kong) for critical reading of the manuscript and the reviewers for their constructive comments on an earlier version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Lightfoot.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Guo, Y., Qiu, LJ. Allele-specific marker development and selection efficiencies for both flavonoid 3′-hydroxylase and flavonoid 3′,5′-hydroxylase genes in soybean subgenus soja . Theor Appl Genet 126, 1445–1455 (2013). https://doi.org/10.1007/s00122-013-2063-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-013-2063-3