
Hypoxia-inducible factor (HIF) is an evolutionarily conserved heterodimeric transcription factor that responds to low oxygen concentrations. The HIF system serves as the oxygen sensor. For its basic research award, 2016, the Albert and Mary Lasker foundation picked the work of William Kaelin Jr., Peter Ratcliffe, and our own Editor-in-Chief, Gregg Semenza, for their contributions to oxygen sensing. The HIF signaling cascade mediates the effects of the state of low oxygen tension within the cell. An obvious HIF primary function is erythropoiesis regulation. Hypoxia often keeps cells from differentiating. Nevertheless, hypoxia promotes the formation of blood vessels and is important for the formation of a vascular system in embryos and tumors. The hypoxia in wounds also promotes the migration of keratinocytes and the restoration of the epithelium. The HIF system is largely regulated by the actions of prolyl hydroxylases (PHDs) [1]. These enzymes hydroxylate specific prolines marking HIF to be targeted for degradation (Fig. 1). PHDs have become drug targets and numerous PHD inhibitors have been developed to treat anemia, ischemia, and even neurological conditions, including Alzheimer disease, Parkinson disease, and Huntington disease [2]. HIF-1α mediates a broad, evolutionarily conserved, endogenous adaptive program to hypoxia and manipulation of HIF pathway components could become organ protective, at least so it is hoped. For most living beings, oxygen is one of the essential elements required to sustain life; however, evolutionarily we were not here first. Anaerobic organisms were first in that regard and oxygen came later.

HIF is a heterodimer, consisting of a constitutively stable HIFβ subunit (also known as aryl hydrocarbon translocator, or ARNT) and an oxygen-sensitive HIFα subunit. HIFα and HIFβ are continuously transcribed and translated [1]. However, under normal oxygen conditions (21% O2), the HIFα subunit is hydroxylated at conserved proline residues in the oxygen-dependent degradation domain by prolyl hydroxylate enzymes (PHDs). The hydroxylated proline residues of the HIFα protein are then recognized by an E3 ubiquitin ligase, von Hippel–Lindau protein (pVHL), which subsequently targets HIF for rapid degradation by the proteasome. The factor-inhibiting HIF (FIH), in addition, hydroxylates HIF at an asparagine residue
Tissue hypoxia is prominent in the micro-environment involved in infections [3]. An important body of evidence suggests that hypoxia and inflammation are interconnected. HIF activation depends on the nature of the pathogen and the characteristics of infections in certain hosts. Currently, whether or not the phenomenon of HIF-1 activation in infections has a protective or detrimental effect on the host is not entirely clear. However, one thing is nonetheless apparent; HIF plays an important role in inflammation [3]. The inflammatory cytokine IL-1β [4] and NO [5] induce HIF activity and HIF activates transcription of the genes encoding iNOS [6] and PHD3 [7]. NO production is tightly linked to oxygen homeostasis. NO synthase isoforms require oxygen. Ho et al. emphasized that HIF stabilization and transcriptional activity is achieved through two parallel pathways: first, a decrease in oxygen-dependent prolyl hydroxylation of HIF and second, the S-nitrosylation of HIF pathway components [8]. NO is one of the few gaseous signaling molecules known and is additionally exceptional as a radical gas. NO is a biological product in almost all types of organisms, ranging from bacteria to plants, fungi, and animal cells [9]. Hypoxia stabilizes HIFs in macrophages and neutrophils and thus serves to regulate inflammatory responses in myeloid cells. Thereby, HIF could shape macrophage and neutrophil function in mediating inflammatory disease [10]. Other HIF stabilization-driven functions in gene expression could influence perfusion and endothelial function, reprogram metabolism to promote glucose intake and glycolysis over oxidative metabolism, reduce inflammation, and beneficially modify the innate immune system [11]. As a matter of fact, evidence indicates that reactive oxygen species (ROS) generated by mitochondrial complex III are required for hypoxic activation of HIF [1].
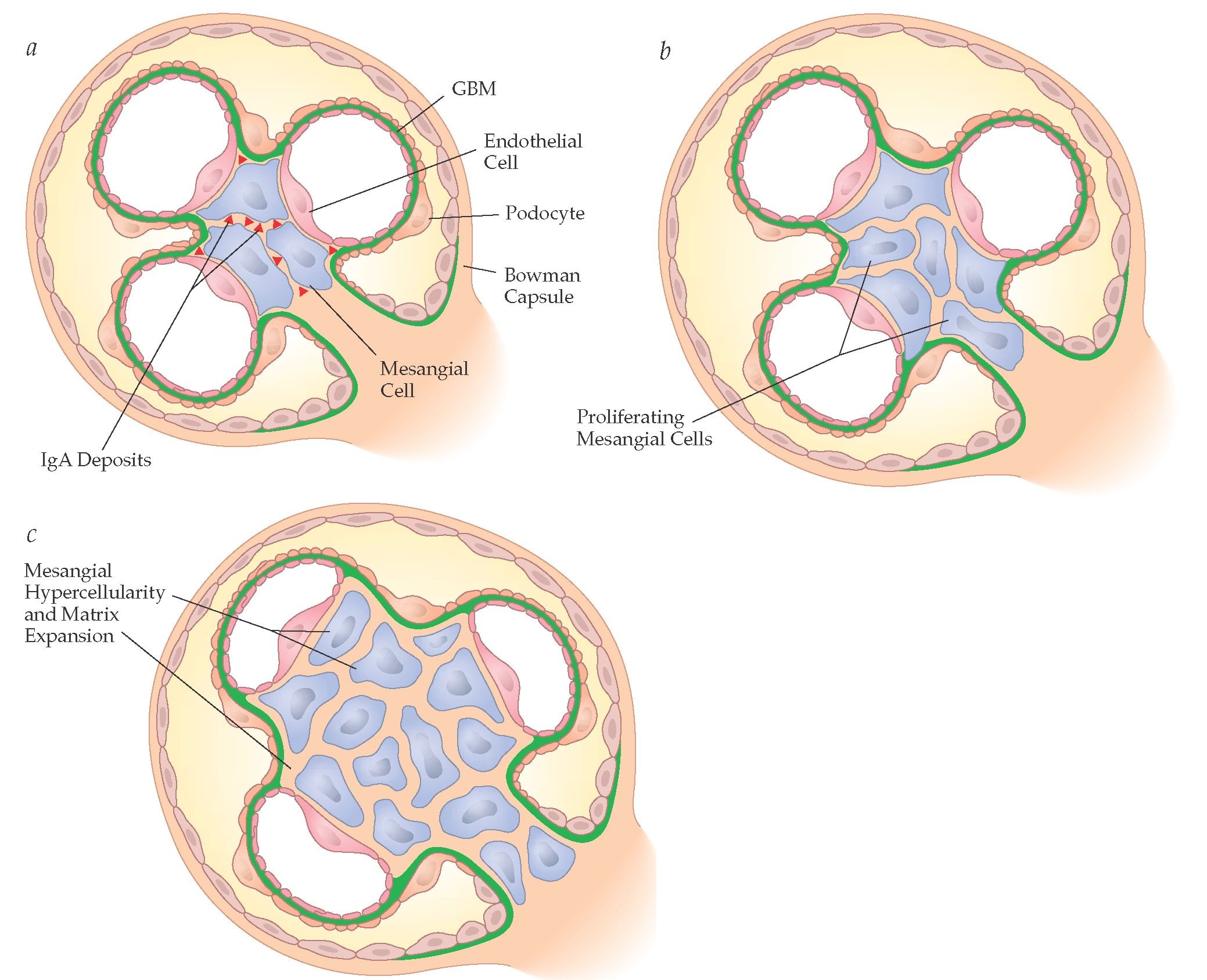
Mesangial cells are specialized cells that largely form the vascular pole of the glomerulus, the kidney’s filtering organ (Fig. 2). The cells make up the renal mesangium within the mesangial matrix. The mesangium is located largely between afferent and efferent arterioles. The putative function of the mesangium is the disposal and degradation of filtration residues and aggregated proteins from the glomerular basement membrane, thereby keeping the glomerulus free of debris. The mesangium also has contractile properties and may thereby contribute to filtration regulation. Mesangial cells are believed to be highly important to glomerular inflammatory diseases [12]. For instance, leukocyte migration and infiltration into the glomerulus is responsible for the initiation and amplification of glomerular injury. Adhesion molecules and chemokines, which can be locally synthesized by mesangial cells, direct the process. Under these circumstances, mesangial cells may proliferate and increase their matrix production. Regulatory mechanisms of mesangial cell replication include a complex array of factors that control cell proliferation, survival, and apoptosis. NO could be a participant that affects mesangial cell adhesion, growth, and survival [13].

Panel a shows a glomerulus (Bowman capsule and contents) with three capillary loops. Glomerular basement membrane (GBM) is in green, podocytes are on the outside while fenestrated endothelial cells are on the inside of the loops. The mesangial cells make up the central supporting structure. The site of IgA deposits are shown. Panel b illustrates what happens when mesangial cells begin to proliferate. Panel c shows considerable mesangial cell proliferation as occurs in both IgA nephropathy and in the anti-Thy1 nephritis model (http://what-when-how.com/wp-content/uploads/2012/04/tmp8954.jpg)
In this issue of J Mol Med, Aglan et al. report that NO mediates PHD3 expression in mesangial cells and in glomerulonephritis [14]. The authors stimulated mouse mesangial cells with a cytokine cocktail including interleukin-1β (IL-1β), tumor necrosis factor–α (TNF-α), lipopolysaccharide (LPS), and interferon-γ (INF-γ). They observed a 60-fold PHD3 and a modest PHD2 induction with this cytokine configuration that they call cytokine-mix-plus (CM+), as opposed to just applying IL-1β and TNF-α alone that they call cytokine mix (CM). CM+ induced NO synthase, which accounted for the massive PHD3 induction, while CM did to a far lesser degree. That the induction might involve nuclear factor kappa-B (NF-κB) comes as no great surprise, accounting for upregulation of numerous NF-κB targets. However, although CM+ also involved NF-κB and its targets, the NO response and HIF-1α upregulation were the more prominent features. The authors showed that the NO response accounts for the PHD3 upregulation.
Thymocyte antigen 1(Thy1) or cluster of differentiation-90 (CD90) is a heavily N-glycosylated glycophosphatidyl-inositol (GPI) anchored cell surface protein. Renal mesangial cells express Thy1. The function of Thy1 is still not fully explained; however, the surface protein has been implicated in cell-cell and cell-matrix interactions, neurite outgrowth, nerve regeneration, apoptosis, metastasis, fibrosis, and inflammation. Thy1 antibody injection is a favorite animal model of glomerulonephritis [15]. Aglan and colleagues used this model, to mimic mesangio-proliferative glomerulonephritis [14]. They observed that PHD3 mRNA and protein expression were induced. An inducible NO-synthase inhibitor ameliorated the findings. Finally, the authors moved to rat mesangial cells to solidify their findings. The cells were treated with IL-1β, which in these cells was sufficient to induce NO synthase. PHD3 expression is indeed increased, an effect that could be blocked with NO synthase inhibition. But can these findings be expanded to human disease?
The anti-Thy1 nephritis model is incomplete. The authors do not report on the course of the model; not even a marker of glomerular filtration rate is reported. The relevance of CD90 to any human renal disease has not been reported. Probably the most important human mesangial glomerular disease is IgA nephropathy [16]. There is no standardized animal model of utility; however, new transgenic models could modify that state-of-affairs. Underglycosylated IgA1 and autoantibodies to these IgA glycoforms and IgA receptors such as CD89 have been implicated. Secondary effector mechanisms, such as investigated by Aglan et al. [17] are important in this disease. These include complement activation and release of various mesangial growth factors are implicated here. The HIF-NO axis and PHD3 have not been studied in IgA nephropathy but should be. The disease is sufficiently common and severe enough that a rigorous search should be made. As a matter of fact, human biopsy tissue specimens are currently available for analysis [17].
Respectfully
Friedrich C. Luft
References
Klimova T, Chandel NS (2008) Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ 15:660–666
Speer RE, Karuppagounder SS, Basso M, Sleiman SF, Kumar A, Brand D, Smirnova N, Gazaryan I, Khim SJ, Ratan RR (2013) Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: from ferroptosis to stroke. Free Radic Biol Med 62:26–36
Devraj G, Beerlage C, Brune B, Kempf VA (2016) Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect. doi:10.1016/j.micinf.2016.11.003
Hellwig-Bürgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W (1999) Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 94(5):1561–1567
Kimura H, Weisz A, Kurashima Y, Hashimoto K, Ogura T, D’Acquisto F, Addeo R, Makuuchi M, Esumi H (2000) Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood 95(1):189–197
Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E (2004) Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J 381(Pt 3):761–767
Palmer LA, Semenza GL, Stoler MH, Johns RA (1998) Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1. Am J Phys 274(2 Pt 1):L212–L219
Ho JJ, Man HS, Marsden PA (2012) Nitric oxide signaling in hypoxia. J Mol Med (Berl) 90:217–231
Roszer T, Banfalvi G (2012) FMRFamide-related peptides: anti-opiate transmitters acting in apoptosis. Peptides 34:177–185
Lin N, Simon MC (2016) Hypoxia-inducible factors: key regulators of myeloid cells during inflammation. J Clin Invest 126:3661–3671
Koivunen P, Serpi R, Dimova EY (2016) Hypoxia-inducible factor prolyl 4-hydroxylase inhibition in cardiometabolic diseases. Pharmacol Res 114:265–273
Gomez-Guerrero C, Hernandez-Vargas P, Lopez-Franco O, Ortiz-Munoz G, Egido J (2005) Mesangial cells and glomerular inflammation: from the pathogenesis to novel therapeutic approaches. Curr Drug Targets Inflamm Allergy 4:341–351
Schaefer L, Beck KF, Raslik I, Walpen S, Mihalik D, Micegova M, Macakova K, Schonherr E, Seidler DG, Varga G et al (2003) Biglycan, a nitric oxide-regulated gene, affects adhesion, growth, and survival of mesangial cells. J Biol Chem 278:26227–26237
Aglan A, Longen S, Dehne N, Köhler Y, Hassan M, Beck M, Tredup C, Boosen M, Hsieh TL, Schaefer L et al (2016) Nitric oxide mediates prolyl hydroxylase 3 expression in mesangial cells and in glomerulonephritis. J Mol Med (Berl) this issue. doi:10.1007/s00109-016-1503-3
Yamamoto T, Wilson CB (1987) Quantitative and qualitative studies of antibody-induced mesangial cell damage in the rat. Kidney Int 32:514–525
Floege J, Moura IC, Daha MR (2014) New insights into the pathogenesis of IgA nephropathy. Semin Immunopathol 36:431–442
Mariani LH, Pendergraft WF 3rd, Kretzler M (2016) Defining glomerular disease in mechanistic terms: implementing an integrative biology approach in nephrology. Clin J Am Soc Nephrol 11:2054–2060
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

{kind=link}
Cite this article
Luft, F.C. Mesangial renal disease, oxygen sensing, and prolyl hydroxylase. J Mol Med 95, 235–237 (2017). https://doi.org/10.1007/s00109-017-1514-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-017-1514-8