
Abstract
The gastrointestinal mucosa is a richly perfused vascular bed directly juxtaposed with the anaerobic and nonsterile lumen of the gut. As such, intestinal epithelial cells, which line the mucosa, experience a uniquely steep physiologic oxygen gradient in comparison with other cells of the body. Inflammation associated with a loss of epithelial barrier function and unregulated exposure of the mucosal immune system to luminal antigens leads to inflammatory bowel disease (IBD), a relatively common disorder with severe morbidity and a limited therapeutic repertoire. During IBD, increased tissue metabolism and vasculitis renders the chronically inflamed mucosa and particularly the epithelium hypoxic, giving rise to the activation of the hypoxia-responsive transcription factor hypoxia-inducible factor (HIF). Recent studies utilizing conditional intestinal epithelial hif1a-null mice have revealed a protective role for epithelial HIF-1α in murine models of IBD. Such protection occurs, at least in part, through HIF-dependent induction of barrier-protective genes in the epithelium. More recently, studies employing pharmacologic activation of HIF via inhibition of HIF prolyl hydroxylases revealed a profoundly protective effect of these agents in murine models of colitis. In this paper, we review this pathway in detail and examine the therapeutic potential for targeting HIF hydroxylases in intestinal mucosal inflammatory disease.
Similar content being viewed by others

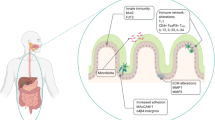

Tissue oxygenation in the gastrointestinal tract
The primary functions of the gastrointestinal tract are the processing and absorption of ingested nutrients, waste removal, fluid homeostasis, and the development of oral tolerance to nonpathogenic luminal antigens. The last of these functions involves the intestinal mucosa being unique among tissues as it is in a constant state of controlled inflammation [1]. This occurs as the mucosal immune system is constantly exposed to new food-borne material in the lumen, which is processed to avoid inappropriate inflammatory reactions to harmless ingested antigens [1].
As well as experiencing this sustained low-grade (physiologic) inflammation, the gut has a unique steady-state tissue oxygenation profile. Firstly, in the physiologic state, the intestinal mucosa experiences multiple daily dynamic fluctuating rates of perfusion. When fasting, a relatively low blood volume is present in the gut; however, after the ingestion of a meal, perfusion rises significantly, resulting in large daily pO2 fluctuations. Secondly, because of its juxtaposition with the anoxic lumen of the gut, the gastrointestinal mucosa has a uniquely steep oxygen gradient from the richly vascularized subepithelial mucosa to the virtually anoxic luminal aspect of the epithelium (Fig. 1). Because of the impressive range of pO2 values that the intestinal mucosa is exposed to on a daily basis, it is perhaps not surprising that resident cells have evolved to be quite resiliant to altered levels of oxygenation.

Mucosal oxygen gradients in normal and inflamed intestinal mucosae. a Under normal physiologic conditions, there exists a steep oxygen gradient across the intestinal mucosa as demonstrated by EF5 staining (red) of colonic epithelial cells (left). Nuclei are stained with DAPI (blue). Tissues from mice treated with TNBS to induce colitis demonstrate dramatically increased EF5 staining reflecting significant inflammation-associated tissue hypoxia. Reproduced in part with copyright permission from the Journal of Clinical Investigation. b Schematic representing mucosal perfusion (red) and inflammatory cell infiltrate (purple) in healthy (left) and inflamed (right) mucosal tissues. Vasculitis and increased inflammatory cell activity combine to cause tissue hypoxia in inflamed tissues
A critical cell type in the maintenance of intestinal homeostasis is the epithelial cell. The intestinal epithelium is a monolayer of cells that covers an area of approximately 250–300 m2 in an adult human and forms a critical barrier between the external (luminal) and internal (vascular) compartments. This dynamic barrier is maintained primarily by the existence of regulated intercellular tight junctions. As well as being a critical barrier, the epithelium is responsible for the absorption of approximately 9 l of fluid from consumed liquids and secreted digestive fluids per day. This fluid transport function is carried out through coordinated ion transport events and the subsequent regulation of salt and water transport between the lumen of the gut and the bloodstream. Importantly, both the barrier and absorptive functions of the intestinal epithelium can be physiologically regulated by oxygen [2–4].
Hypoxia and mucosal inflammation
Inflammatory bowel disease (IBD) is an umbrella term for a range of disorders including ulcerative colitis and Crohn’s disease, which are characterized by a breakdown in the intestinal epithelial barrier with subsequent unregulated exposure of the mucosal immune system to luminal antigenic material leading to inflammation and further barrier breakdown. Thus, a self-perpetuating cycle of inflammation is initiated leading to severe pathology [5–7]. Because of the limited number of current therapeutic options available, treatment often ultimately resorts to surgical resection of significant amounts of chronically inflamed intestinal tissue.
Active inflammation is characterized by dramatic shifts in tissue metabolism and perfusion. These changes include diminished availability of oxygen (hypoxia) [8–10] with subsequent lactate accumulation and resultant metabolic acidosis. Such shifts in tissue metabolism result, at least in part, from profound recruitment of inflammatory cells, in particular myeloid cells such as neutrophils (polymorphonuclear cells) and monocytes. The vast majority of inflammatory cells are not resident cells but are recruited to inflammatory lesions [11]. As such, it is important to understand the interactions between microenvironmental metabolic changes (e.g., hypoxia) as they relate to molecular mechanisms of leukocyte recruitment and intestinal epithelial dysfunction during inflammation. More importantly, it is imperative to define whether mechanisms initiated by hypoxia might serve as potential therapeutic targets.
A number of studies have implicated the occurrence of hypoxia in mucosal inflammatory diseases such as IBD [12]. Surgical specimens from patients with IBD have revealed prominent hypoxia-inducible factor (HIF)-1 and HIF-2 activation associated with increased vascular density in diseased areas [13]. Other studies in humans have revealed that a number of microvascular abnormalities may contribute to diminished blood flow to the intestine in IBD, including the loss of endothelial nitric oxide generation and enhanced tissue vasoconstrictor production [12]. Moreover, Vascular endothelial growth factor-dependent angiogenesis appears to be an integral part of human IBD [14]. In support of these hypotheses, studies in murine models have identified the epithelium as the central target of hypoxia during active mucosal inflammation [15]. As part of our ongoing work, we have confirmed the existence of mucosal hypoxia in murine models of IBD using 2-nitroimidazole dyes, a class of compounds known to undergo intracellular metabolism depending on the availability of oxygen within tissue (Fig. 1). Nitroimidazoles enter viable cells where they undergo a single electron reduction, to form a reactive intermediate species. In the presence of normal oxygen levels, the molecule is immediately reoxidized and diffuses out of the cell. In the absence of adequate oxygen concentrations, the molecule is incompletely reoxidized, and the highly reactive reduced form associates with intracellular proteins, forming adducts that can be localized with antibodies [16].
Localization of hypoxia utilizing these 2-nitroimidazole dyes revealed two interesting observations. First, in the small intestine and especially the colon, “physiologic hypoxia” appears to predominate. Indeed, accumulation of nitroimidazole adducts were readily evident in epithelial cells lining the lumenal aspect of the intestine. This was not the case in other tissues (e.g., lung and liver, unpublished observation), confirming previous studies that the resting pO2 in the intestinal epithelium is quite low, likely because of the steep gradient of oxygen across the lumenal aspect. Second, these imaging studies revealed that cells overlying mucosal lesions are considerably more hypoxic. Accumulation of nitroimidazole adducts, particularly in the epithelium, were as intense as those observed in some tumors, suggesting the existence of intense foci of hypoxia associated with these inflammatory lesions. While we do not yet know the basis for such inflammatory hypoxia, some evidence suggests that tissue vasculitis could predispose epithelia toward diminished oxygen delivery [15].
HIF is protective for mucosal inflammation
A number of studies have revealed that HIF elicits a barrier protective program in the intestine [17–20]. While originally guided by microarray analysis of differentially expressed messenger ribonucleic acid (mRNA) in cultured epithelial cells subjected to hypoxia, these studies have proven robust in a number of animal models of inflammation. Further interrogation of mechanisms related to hypoxia-elicted barrier protection have revealed three important features. First, expression of the functional proteins encoded by these mRNAs was localized to the most lumenal aspect of polarized epithelia (i.e., apically expressed proteins). Second, molecular dissection of the hypoxia-elicited pathway(s) for this “apical gene cluster” revealed a high propensity for regulation by HIF. Third, HIF-dependent epithelial barrier-protective pathways driven by hypoxia tend to be more “nonclassical” regulators of barrier function. Rather than classic junctional proteins such as occludin or claudin(s), hypoxia-induced enhancement of barrier function occurs through diverse pathways, ranging from increased mucin production [21] and molecules that modify mucins (e.g., intestinal trefoil factor) [17], to xenobiotic clearance (P-glycoprotein) [18] to nucleotide metabolism (ecto-5′-nucleotidase, CD73) [19–20] and nucleotide signaling (adenosine A2B receptor) [20] (Fig. 2).

HIF-dependent barrier protective gene expression in intestinal epithelial cells. Under conditions of hypoxia, intestinal epithelial cells express a number of barrier protective genes in a HIF-1 dependent manner
To more fully understand the physiologic implications of intestinal epithelial HIF, Karhausen et al. [15] generated two mouse lines with intestinal epithelial-targeted expression of either mutant Hif1a (constitutive repression of HIF-1) or mutant von Hippel-Lindau gene (Vhlh, constitutive overexpression of HIF, which includes HIF-1 and HIF-2). Studies of colitis in these mice revealed that the loss of epithelial HIF-1 correlated with more severe clinical symptoms (mortality, weight loss, colon length, intestinal epithelial permeability), whereas an increase in epithelial HIF was protective for these individual parameters. These studies clearly demonstrated that HIF-1α plays a critical role in barrier maintenance and provide evidence for our initial hypothesis of a HIF-1-controlled apical gene cluster. The role of HIF-2α in inflammatory lesions in the intestine remains less clear. However, given the differences in both tissue distribution patterns and target gene preferences between HIF-1α and HIF-2α, it is likely that this isoform plays a distinct role in IBD. Future studies will address this important question.
Further evidence in support of a protective role for HIF in mucosal disease are provided by studies directed at HIF prolyl hydroxylase (PHD) inhibitors [22, 23]. These enzymes were identified on the principle that other mammalian PHDs such as those which target extracellular collagen were 2-oxoglutarate dependent [24], and it was predicted that the HIF PHDs would also belong to this family of enzymes. Based on conserved structural features [24], a candidate molecular approach was used to define HIF-modifying enzymes. This approach identified the HIF PHDs as the products of genes related to C.elegans egl-9, a gene that was first described in the context of an egg-laying abnormal phenotype [25]. In mammalian cells, three PHD isoforms were identified (PHD 1–3), and shown to hydroxylate HIF-α in vitro [26–27]. These enzymes have an absolute requirement for oxygen as the substrate. The overall reaction results in insertion of one oxygen atom into the HIF-α peptide substrate at the proline residue, with the other oxygen molecule generating succinate from 2-OG with the release of CO2. Reactions conducted in a limited oxygen environment have revealed that the activity of the purified enzyme is strikingly sensitive to diminished levels of oxygen in vitro [26–27]. The three enzymes have different tissue distributions and, at least under conditions of overexpression, have distinct patterns of subcellular localization [15, 19]. PHD1 mRNA is expressed in many tissues, with especially high expression in the testis. Likewise, PHD2 mRNA is widely expressed, with particularly abundant expression in adipose tissue [24, 28]. PHD3 mRNA is also expressed in many tissues but is most abundant in the heart and placenta [24, 28]. In mouse intestinal mucosal tissue, we have found expression all three isoforms of PHDs with a distribution of PHD1 < PHD2 = PHD3 [22–23].
The discovery of HIF-selective PHDs as central regulators of HIF expression has now provided the basis for potential development of PHD-based molecular tools and therapies [29–30]. Pharmacological inactivation of the PHDs by 2-OG analogues is sufficient to stabilize HIF-α [29], but this action is nonspecific with respect to individual PHD isoforms. In vitro studies suggest significant differences in substrate specificity. For example, comparison of enzyme activity in vitro showed that the HIF ODD sequence is hydroxylated most efficiently by PHD2 [24, 28]. These observations have generated interest in identifying enzyme-modifying therapeutics. Indeed, a number of PHD inhibitors have been described, including direct inhibitors of the PHDs [31–32], analogs of naturally occurring cyclic hydroxamates [23], as well as antagonists of α-keto-glutarate [29]. As such, we hypothesized that pharmacologic activation of HIF would provide a protective adaptation to murine colitic disease. For these purposes, we have used PHD inhibitors that stabilize HIF-α and subsequently drive the expression of downstream HIF target genes. Our results show that the PHD inhibition provides an overall beneficial influence on clinical symptoms (weight loss, colon length, tissue tumor necrosis factor-α/interferon-γ) in multiple murine models of colitis. These effects are most likely due to their barrier-protective function and enhancement of wound healing at the site of inflammation [22–23]. Taken together, these findings emphasize the role of epithelial HIF-1α during inflammatory diseases in the colon and may provide the basis for a therapeutic use of PHD inhibitors in inflammatory mucosal disease.
Critically, HIF is not the only hypoxia-responsive transcription factor, and the oxygen-dependent regulatory role of hydroxylases is not be restricted to HIF [33]. Indeed, recent studies have indicated that the nuclear factor (NF) κB pathway may also be regulated in a similar manner. Hypoxia activates NF-κB, and this appears at least in part to be mediated through altered hydroxylation of critical components of this pathway [33–34]. It is interesting to note that like conditional HIF-1α-null mice, deletion of the NF-κB pathway in intestinal epithelial cells leads to increased susceptibility to colitis indicating a protective role for epithelial NF-κB in colitis. This effect is likely mediated through increased expression of antiapoptotic genes in the intestinal epithelium resulting in enhanced epithelial barrier function. Thus, a significant part of the protective effect of hydroxylase inhibition in models of colitis may be through the promotion of intestinal epithelial NF-κB activity [22] (Fig. 3). Ongoing studies using conditional knockout mice are investigating the relative importance of the HIF and NF-κB pathways in determining the protective effects of hydroxylase inhibition in colitis.

Hypoxia-dependent HIF and NF-κB activation in intestinal epithelial cells. Exposure of cells to hypoxia or the hydroxylase inhibitor DMOG results in hydroxylase inhibition, which facilitates activation of both the HIF and NF-κB pathways. HIF-1-dependent pathways lead to enhanced epithelial barrier function through the expression of barrier protective genes. NF-κB likely enhances barrier function by the prevention of apoptosis of intestinal epithelial cells. In concert, these two pathways effectively increase barrier function and are thus protective against colitis
Signaling interactions between hypoxia and inflammation
As outlined above, both the HIF and NF-κB pathways are activated under conditions of hypoxia. While the role of hydroxylases in the hypoxic sensitivity of the HIF pathway has been clearly demonstrated, recent data raises the intriguing possibility that components of the NF-κB pathway may also be substrates of hydroxylases including PHD1 and FIH [33–34]. It is interesting to note that as well as being hypoxia sensitive, both the HIF and NF-κB pathways are regulated by inflammatory mediators including cytokines and bacterial products such as lippopolysaccharide [35–36] (Fig. 4). A range of inflammatory stimuli activate NF-κB through receptor occupation and activation of a complex and diverse array of receptor specific signal transduction pathways. Critically, one of the gene targets of NF-κB is HIF-1α. Thus, inflammatory stimuli activate the HIF pathway through transcriptional upregulation of the HIF-1 mRNA expression in an NF-κB-dependent manner. Conversely, NF-κB activity in hypoxia can be regulated by HIF [37]. Clearly, an intimate relationship exists between NF-κB and HIF-1 signaling in the context of microenvironments where hypoxia and inflammation coexist such as the inflamed bowel. Intestinal epithelial cells are unique in that they are constantly exposed to inflammatory stimuli and a steep oxygen gradient, which may underscore the importance of these pathways in the regulation of epithelial cell function both in physiology and disease.

Interactions between HIF and NF-κB signaling pathways. Both HIF and NF-κB are activated in hypoxia through decreased hydroxylase activity. Similarly, both HIF-1 and NF-κB are activated by proinflammatory mediators such as cytokines and bacterial lipopolysaccharide. It is interesting to note that NF-κB activates transcriptional upregulation of HIF-1a mRNA indicating one level at which these two pathways interact to regulate hypoxia-dependent gene transcription
Conclusions and perspectives
The gastrointestinal mucosa provides a unique setting to study tissue oxygenation and changes in disease states. The relatively low baseline pO2 coupled with high blood flow and energy demand against a background of physiologic inflammatory activity identify this mucosal surface as having high potential for targeted HIF-based therapy. Results from animal models of IBD have demonstrated an overall beneficial impact of hydroxylase inhibition. Key issues remaining to be elucidated include identification of the critical gene targets involved, determination of the relative roles of HIF and NF-κB pathways, identification of tissue-specific expression of HIF PHD isoforms, and elucidation of the role of HIF-2α in this protective response. In summary, the endogenous adaptive pathways activated in response to hypoxia represent potentially important new windows of therapeutic opportunity in IBD.
References
Poonam P (2007) The biology of oral tolerance and issues related to oral vaccine design. Curr Pharm Des 13:2001–2007
Friedman GB, Taylor CT, Parkos CA, Colgan SP (1998) Epithelial permeability induced by neutrophil transmigration is potentiated by hypoxia: role of intracellular cAMP. J Cell Physiol 176:76–84
Taylor CT, Dzus AL, Colgan SP (1998) Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology 114:657–668
Taylor CT, Lisco SJ, Awtrey CS, Colgan SP (1998) Hypoxia inhibits cyclic nucleotide-stimulated epithelial ion transport: role for nucleotide cyclases as oxygen sensors. J Pharmacol Exp Ther 284:568–575
Xavier RJ, Podolsky DK (2007) Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–434
Baumgart DC, Carding SR (2007) Inflammatory bowel disease: cause and immunobiology. Lancet 369:1627–1640
Sartor RB (1995) Current concepts of the aetiology and pathogenesis of ulcerative colitis and Crohn's disease. Gastroenterol Clin North Am 24:475–507
Haddad JJ (2003) Science review: redox and oxygen-sensitive transcription factors in the regulation of oxidant-mediated lung injury: role for hypoxia-inducible factor-1alpha. Crit Care 7:47–54
Kokura S, Yoshida N, Yoshikawa T (2002) Anoxia/reoxygenation-induced leukocyte–endothelial cell interactions. Free Radic Biol Med 33:427–432
Saadi S, Wrenshall LE, Platt J (2003) Regional manifestations and control of the immune system. FASEB J 16:849–856
Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE (1999) Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol 66:889–900
Hatoum OA, Binion DG, Gutterman DD (2005) Paradox of simultaneous intestinal ischaemia and hyperaemia in inflammatory bowel disease. Eur J Clin Invest 35:599–609
Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter KC, Harris AL, Koukourakis MI (2003) Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol 56:209–213
Danese S, Dejana E, Fiocchi C (2007) Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J Immunol 178:6017–6022
Karhausen JO, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH (2004) Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 114:1098–1106
Evans SM, Hahn S, Pook DR, Jenkins WT, Chalian AA, Zhang P, Stevens C, Weber R, Weinstein G, Benjamin I, Mirza N, Morgan M, Rubin S, McKenna WG, Lord EM, Koch CJ (2000) Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res 60:2018–2024
Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford KM, Narravula S, Podolsky DK, Colgan SP (2001) Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 193:1027–1034
Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP (2002) Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 62:3387–3394
Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP (2002) Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 (HIF-1) mediates permeability changes in intestinal epithelia. J Clin Invest 110:993–1002
Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP (2003) Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. Exp Med 198:783–796
Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP (2006) Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem 99:1616–1627
Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PF, Taylor CT (2008) The hydroxylase inhibitor DMOG is protective in a murine model of colitis. Gastroenterol (in press)
Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP (2008) Mucosal protection by HIF prolyl hydroxylase inhibition. Gastroenterology (in press)
Bruick RK (2003) Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev 17:2614–2623
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107:43–54
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472
Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY (2002) Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 417:975–978
Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5:343–354
Mole DR, Schlemminger I, McNeill LA, Hewitson KS, Pugh CW, Ratcliffe PJ, Schofield CJ (2003) 2-oxoglutarate analogue inhibitors of HIF prolyl hydroxylase. Bioorg Med Chem Lett 13:2677–2680
Masson N, Ratcliffe PJ (2003) HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O(2) levels. J Cell Sci 116:3041–3049
Nwogu JI, Geenen D, Bean M, Brenner MC, Huang X, Buttrick PM (2001) Inhibition of collagen synthesis with prolyl 4-hydroxylase inhibitor improves left ventricular function and alters the pattern of left ventricular dilatation after myocardial infarction. Circulation 104:2216–2221
Schlemminger I, Mole DR, McNeill LA, Dhanda A, Hewitson KS, Tian YM, Ratcliffe PJ, Pugh CW, Schofield CJ (2003) Analogues of dealanylalahopcin are inhibitors of human HIF prolyl hydroxylases. Bioorg Med Chem Lett 13:1451–1454
Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT (2006) Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 103:18154–18159
Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe PJ (2006) Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci USA 103:14767–14772
Pouyssegur J, Mechta-Grigoriou F (2006) Redox regulation of the hypoxia-inducible factor. Biol Chem 387:1337–1346
Frede S, Stockmann C, Freitag P, Fandrey J (2006) Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p42/44 MAPK and NF-kappaB. Biochem J 396:517–527
Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med 201:105–15
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Taylor, C.T., Colgan, S.P. Hypoxia and gastrointestinal disease. J Mol Med 85, 1295–1300 (2007). https://doi.org/10.1007/s00109-007-0277-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-007-0277-z