
Abstract
In absence of experimental 3D structures, several homology models, based on ABC exporter 3D structures, have provided significant insights into the molecular mechanisms underlying the function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, a chloride channel whose defects are associated with cystic fibrosis (CF). Until now, these models, however, did not furnished much insights into the continuous way that ions could follow from the cytosol to the extracellular milieu in the open form of the channel. Here, we have built a refined model of CFTR, based on the outward-facing Sav1866 experimental 3D structure and integrating the evolutionary and structural information available today. Molecular dynamics simulations revealed significant conformational changes, resulting in a full-open channel, accessible from the cytosol through lateral tunnels displayed in the long intracellular loops (ICLs). At the same time, the region of nucleotide-binding domain 1 in contact with one of the ICLs and carrying amino acid F508, the deletion of which is the most common CF-causing mutation, was found to adopt an alternative but stable position. Then, in a second step, this first stable full-open conformation evolved toward another stable state, in which only a limited displacement of the upper part of the transmembrane helices leads to a closure of the channel, in a conformation very close to that adopted by the Atm1 ABC exporter, in an inward-facing conformation. These models, supported by experimental data, provide significant new insights into the CFTR structure–function relationships and into the possible impact of CF-causing mutations.
Similar content being viewed by others


Introduction
Cystic fibrosis transmembrane conductance regulator (CFTR) is a member of the ATP-binding cassette (ABCC7) family of transmembrane (TM) proteins that functions as a phosphorylation-regulated, ATP-gated Cl− channel [1, 2]. Mutations in CFTR cause Cystic Fibrosis (CF), one of the most common inherited disease in the Caucasian population. Over the last years, significant efforts have been made to identify compounds that could restore mutant CFTR function, either by increasing the mutant’s biosynthetic processing efficiency and cell-surface density (correctors) or the channel activity itself (potentiators) [3]. In this context, 3D structure information can be useful not only for getting insights into the molecular basis of CFTR function but also for understanding, at the molecular level, the mechanism of action of compounds that would directly target the mutated CFTR protein, as well as for identifying new CFTR-specific active compounds. CFTR, like the other members of the ABC exporter family to which it belongs, is composed of two membrane-spanning domains (MSDs), consisting each of six helices, and two soluble nucleotide-binding domains (NBDs). As learned from experimental 3D structures [4], the particularity of ABC exporters, versus importers, is that the TM helices do largely protrude (~42 Å) into the cytoplasm and thereby form long intracellular loops (ICLs). The tips of the ICLs are made of short coupling helices, running parallel to the membrane plane and making transmission interfaces with the NBDs. CFTR is, however, a unique case among ABC exporters as it is the only known member that behaves as an anion channel, whereas ABC exporters are generally involved in the transport of much larger substrates. CFTR channel pore opening is mediated by the phosphorylation of its large cytoplasmic regulatory (R) domain and by ATP binding at the NBDs, which induce conformational changes within the MSDs allowing anions to flow. Probably, the CFTR cycle involves complex sequences of multiple steps, associated with different sub-conductance states and leading then to a stable, full-open state of the channel [5].
In the absence of experimental 3D structures at atomic resolution for the entire CFTR protein, homology models have been built for the MSD–NBD assembly, providing significant insights into its specific structural and functional features. The two first models [6, 7] have been constructed using as template the experimental 3D structure of Staphylococcus aureus Sav1866 [8], which was obtained at good resolution (pdb 2hyd, 3.0 Å) in an outward-facing conformation. This conformation was assumed to be a reasonable template for modeling the open form of the CFTR channel, as the two NBDs are tightly associated in a head-to-tail conformation, with the nucleotides bound at the interface between the two NBDs. Here, the ICLs cluster in a compact structure, characterized by a four-helix bundle core, at the basis of which an acido-basic couple plays a key role in the channel function [9]. Subsequently, a model of a possible closed form of the CFTR channel has been constructed [10], using as template the experimental “closed-apo” structure of the inward-facing conformation of Vibrio cholerae MsbA (pdb 3b5x, 5.5 Å) [11]. The comparison of the open and closed CFTR models showed, in particular, that a “ball-and-socket” joint [12], consisting of the ICL2 and ICL4 coupling helices and docking into a cleft of the NBD surface, was maintained during conformational transitions, while the overall four-helix compact ICLs core is lost. The conserved coupling interface contains the amino acid residue Phe508 (F508), the deletion of which is the most common CF-causing mutation and leads to a misfolded protein, which is retained in the endoplasmic reticulum and diverted to degradation by the proteasome [13]. In the models reported until now, the nucleotide-binding domain 1 (NBD1)-located Phe508 makes critical contacts not only with several aromatic amino acids of ICL4 (F1068, Y1073, and F1074), but, most importantly, also with the side chain of R1070. The models thus emphasize the crucial role played by F508 in the interactions of NBD1 with other CFTR domains and stabilizing the whole CFTR assembly [14, 15]. In addition, the absence of F508 has also been demonstrated to alter the thermodynamic stability of NBD1 itself [16, 17]. Accordingly, research now focuses on the identification of combinations of correctors, which would target the multiple conformational defects of F508del [18, 19]. Pharmacological rescue of F508del will probably also require the use of a potentiator, as the mutated F508del protein has abnormal channel gating activity [13].
This initial modeling work has been enriched with a few studies combining modeling and molecular dynamics approaches. These studies, which were based on Sav1866-derived models and focused on the MSD assembly, allowed the description of a possible architecture of the anion conduction path, exhibiting a “hour-glass” shape, with a clear “bottleneck” [20, 21] or a clear narrowing of the pore [22]. A more recent study has also explored the transition from the inward- to outward-facing conformation [23], the starting point being here a mouse P-glycoprotein-derived model [24]. This transition involves a rapid NBD dimerization, driven by the attractive interaction between the ATP adenine ring and the ABC signature motif, with formation of a MsbA “closed-apo”-like conformation. This simulation suggested that ATP-binding constitutes the power stroke which allows the subsequent conformational change in the MSDs. Very recently, another dynamics study investigated the possible conformational changes underlying the gating transition in CFTR, starting from experimentally derived constraints to build 3D models on the Sav1866 and P-glycoprotein templates and using targeted molecular dynamics [25]. Importantly, most of these studies used several experimentally derived data (mainly distance restraints) as constraints to guide the dynamics of the considered models and, consequently, these imposed elements persist in the models finally obtained. Also importantly, these models generally do not show clear entrance to the transmembrane pore from the cytoplasmic end.
Since the initial modeling studies discussed above, several new experimental 3D structures of ABC exporters have been published. All of them concern inward-facing conformations, which were obtained with better resolution than the first 3D structures of MsbA, and in which the separation of the NBDs varies from (1) full or moderate dissociation (P-glycoprotein from mouse: pdb 3g5u (3.80 Å) and 3g60 (4.40 Å) [24], pdb 4ksb (3.80 Å) [26], from Caenorhabditis elegans: pdb 4f4c (3.40 Å) [27], and a P-glycoprotein homolog (ABCB1) from Cyanidioschyzon merolae: pdb 3wme (2.75 Å) and 3wmf (2.40 Å) [28]) to (2) partial dissociation (human ABCB10: pdb 3zdq (2.85 Å); 4ayt (2.85 Å), 4ayx (2.90 Å), and 4ayw (3.30 Å) [29]; Atm1 from yeast: pdb:4myc (3.06 Å) and 4myh (3.38 Å) [30] and from Novosphingobium aromaticivorans: pdb:4mrn (2.50 Å), 4mrp (2.50 Å), 4mrr (297 Å), 4mrs (2.35 Å), and 4mrv ((2.50 Å) [31] and Thermotoga maritima “TM287/288”: pdb 3qf4 (2.9 Å) [32]). The “TM287/288” structure has, as CFTR, asymmetric NBDs, with one degenerate and one consensus nucleotide-binding sites. Accordingly, and in agreement with experimental data obtained for CFTR [33, 34], ATP remains bound to the degenerate site during the entire transport cycle, whereas it is hydrolyzed at the canonical site. While this manuscript was under revision, the experimental 3D structure of yet another, symmetric ABC exporter (McjD) has been published in a new, outward occluded conformation [35].
In the present work, we first considered the whole set of evolutionary and structural information available today, to generate highly accurate sequence alignments and consequently, models with improved accuracy. Such an analysis led to very few but likely important variations in the crucial MSD regions in comparison with our previously published alignment [6, 10]. The Sav1866-based model of the CFTR MSD:NBD architecture, built on this refined alignment and in the absence of any experimental constraint, was further explored using molecular dynamics (MD) simulations, again without considering any experimental constraint. Analysis of the MD frames in several independent replica led to highlight first, very rapidly, a full-open conformer of the CFTR protein, in which the salt bridge between R352 and D993, as reported by Cui et al. [5], is well formed and in which opening of large lateral tunnels may allow the passage of ions and small molecules from the cytosol to the channel. Remarkably, the onset of this full-open conformation is linked to a switch of the NBD1 region bearing F508 toward an alternative position that, to our knowledge, has not yet been observed in ABC exporter 3D structures. This full-open channel model then evolved toward a closed form, after local movement of the upper part of the transmembrane helices, the rest of the structure being essentially unchanged. These results, supported by experimental 3D data, are in favor of relatively localized conformational changes for opening and closing a gate in the transmembrane pore [1] and underline a possible important role of the F508 region in this mechanism.
Materials and methods
Sequence alignments
Sequence alignments have been performed using a combination of standard programs, hydrophobic cluster analysis (HCA) [36, 37] and human expertise. This combination generally plays key role when considering remote relationships (very low levels of sequence identities), for which automated bioinformatics tools, even very sensitive, may lead to locally false alignments. Refinements of alignments are made possible by adding information on the 2D structure of the proteins, which is much more conserved. This strategy has already led to accurately align the sequence of the first β-strand of NBD1 with those of NBDs of other ABC exporters, which was otherwise misaligned using automatic procedures due to the presence of a large regulatory insertion separating it from the rest of the domain [38]. This prediction was supported by the X-ray structure of NBD1 [39]. Here, for the alignment of the MSDs, the strategy includes visual inspection of the superimposed 3D structures, to define the structural invariants, which can be used to guide the alignment of all the sequences of the family in regions where very low level of sequence identity (if any) is observed. The anchor points of the alignments, shown in Online Resource 1, are described in details in Online Resource 2 and examples of the structural analysis are given in Online Resource 3.
Comparative modeling
A model of the MSD:NBD assembly [amino acids 65–649 (MSD1:NBD1), including the regulatory insertion (403–435), and amino acids 845–1,446 (MSD2:NBD2), including the linker insertion (1,182–1,202)], was built using Modeller 9.10 [40], on the basis of the alignments described above. This model (called initial model in the “Results” section) does not include the R domain, for which no valuable 3D template can be found for modeling it with a similar accuracy. The overall stereo-chemical quality of the model was assessed using Procheck [41]. Swiss-PdbViewer [42], Chimera [43], and VMD [44] were used as visualization programs. The POCASA program was used for highlighting internal cavities [45].
MD protocol
The protein was embedded in a lipid bilayer consisting of 319 POPC molecules (160 and 159 lipids in the extracellular and the cytoplasmic leaflets, respectively), and solvated with water molecules and 15 mM NaCl. The resulting simulation system contains about 210,000 atoms. The dimension of the simulation box is 125 × 125 × 170 Å3. The PPM server was used to calculate rotational and translational positions of the protein 3D structure in the membrane [46].
The system has been built using the VMD software [44]. The membrane plugin was used to build the membrane, the Solvate plugin to add the water molecules and the Autoionize plugin to add ions.
The simulation protocol was inspired from [47]. We first performed a short minimization before a 2 ns simulation in which all atoms except lipid tails are kept fixed, to induce the appropriate disorder of a fluid-like bilayer. This step is followed by a 2 ns simulation with the protein fixed, releasing the water, ions, and lipid head groups. As suggested in [47], water molecules were kept outside of the membrane during this phase, for preventing hydration of the membrane–protein interface during equilibration.
These two steps guided the system to the nearest local energy minimum in configuration space, with the protein constrained, allowing the environment to relax first. The protein was then released of any constraints and water molecules were not anymore kept outside the membrane. We performed a short minimization prior to a molecular dynamics of the entire system lasting 10–30 ns.
Molecular dynamics simulations were performed using NAMD 2.9 [48] with the CHARMM22 protein force field [49], including the φ/ψ cross-term map (CMAP) correction for the proteins [50], and CHARMM27 lipid force field [51]. Waters were modeled using TIP3P [52]. Non-bonded and electrostatic interactions were calculated every time steps, with a cutoff of 12 Å. The SHAKE algorithm [53] was used to constrain the lengths of bonds involving hydrogen atoms. The particle mesh Ewald summation [54] was used to calculate Coulomb interactions. Temperature was kept constant using Langevin dynamics and a Nose–Hoover Langevin piston [55] was used for pressure control (NPT, 1 bar and 310 K).
Coordinates of the two models
The coordinates of the two models described here [open and closed forms (conformers 1 and 2, respectively)], as well as those of the initial model, before MD, are available at http://www.impmc.upmc.fr/~callebau/CFTR_2014.html.
Results
Refined alignment considering structural data and evolutionary information, including the symmetry between the two ABC exporter subunits or halves
We compared a large number of sequences from the ABC exporter family using HCA (see [6, 10]), to define structural invariants and thereby refine our alignment in the most divergent regions (i.e., the MSDs) (Online Resource 1). To that aim, we also took into account the symmetry existing between the two different subunits (A and B) of each ABC protein (or the two halves, when the two subunits are fused into a single polypeptidic chain, as in CFTR and P-glycoprotein), except for proteins for which the two subunits are identical (true dimers, shaded gray). We also considered in this alignment the sequences of all the experimental 3D structures of ABC exporters published so far (Staphylococcus aureus Sav1866, T. maritima “TM287/288”, Vibrio cholerae MsbA, mouse P-glycoprotein, C. elegans P-glycoprotein, human ABCB10). The recently published 3D structures of Atm1 from Saccharomyces cerevisiae and N. aromaticivorans and ABCB1 from C. merolae were added afterward. The sequence of E. coli McjD, whose 3D structure was published while this manuscript was under revision [35], can be well aligned with the sequences shown here. Consideration of these experimental information allowed us to assess the reliability of our alignment of the CFTR sequence with those of known 3D structures by comparing the HCA-based alignment of the sequences of these known 3D structures to that directly deduced from their superimposition. In this context, we noted that our alignment perfectly agrees with that recently realized by [28], who used the PROMALS3D program with manual adjustment. This supports the reliability of the alignment of CFTR with all these sequences, sharing similar levels of sequence identity.
As the experimental 3D structures are in different conformations, these were compared through a rigid block approach, which considered, as independent structural elements, sequence stretches for which the 3D structure as a whole does not dramatically change between the different conformations (outward- and inward-facing conformations). Thus, these rigid blocks either correspond to whole domains (NBDs) or limited parts of domains (MSDs), only including a few regular secondary structures. Hence, we observed that the MSDs can be divided in four blocks of three consecutive helices, based on an internal symmetry we detected within these domains (Online Resource 4), i.e., TM1–3 (Block 1, highlighted in blue), TM4–6 (Block 2, green), TM7–9 (Block 3, yellow), and TM10–12 (Block 4, red). Remarkably, the conformational transition between extreme (outward- and inward-facing) conformations of ABC exporters can be modeled by rotation of these four rigid three-helix blocks (Online Resource 4). Such a kind of analysis of the alternating conformations of ABC exporters in terms of structurally conserved elements has already been performed by other groups, but considering different blocks (TM4–5 and TM1–2–3/6 [11] and TM1–2, TM4–5, and TM3/TM6 [31]).
The structural analysis of positions for which striking amino acid conservation is observed (boxed and colored) is detailed in Online Resource 2. A few of these positions are illustrated in Online Resource 3. Outside the NBDs, the coupling helices of the four ICLs obviously constitute strong anchor points for assessing the reliability of the alignment. A limited number of insertions/deletions are observed in MSDs, but their precise positions are particularly hard to define (Online Resource 1). Relative to our previous alignment [6, 10], there are only a few but important differences which are as follows: (1) the N-helix (amino acids 65–76) is shifted upstream (toward the N terminus) by four positions, (2) the presence, in Sav1866 TM2, of two consecutive proline residues (amino acids 75 and 76) induces upstream a π-turn (R74 NH···I69 O, H-bond of 2.94 Å). Thus, relative to all the other sequences of the family, an insertion has to be made in the Sav1866 sequence at position 69 [Online Resource 3(A)]. Accordingly, the CFTR segments 113–129 (TM2) and 899–923 (TM8) are also shifted upstream by one position. However, of note, such a shift does not systematically occur when there are two consecutive proline residues within a α-helix (e.g., positions 164–165 in “TM287”, 264–265 in “TM288”, 740–741 in mouse P-gp, 1,009–1,010 in C. elegans P-gp, and 323–324 in ABCB10).
To allow comparison, Online Resource 5(A) illustrates the main characteristics of models of the CFTR MSD:NBD assembly that were previously published, including those which were analyzed by MD studies. Online Resource 5(B) indicates the shifts observed between the corresponding sequence alignments. In the MSDs, the differences range from 0 to 8 amino acids, depending on the considered secondary structure. Although relatively small, they may lead to large 3D discrepancies, especially due to a ~90° rotation per amino acid along the entire considered long alpha-helical path. As noted before, our alignment was remarkably in agreement with the structural alignment of ABC exporters reported recently [28] and differs by only one deletion introduced in TM2 by the P75–P76 motif (see above).
This consolidated alignment was next used as the basis for reconstructing a new homology model of the CFTR MSD:NBD assembly, without the R domain for which no 3D structure is available to model it with a similar accuracy. Two ABC exporter structures (Sav1866 and MsbA) are now available in a highly similar outward-facing conformation. Here again, we used Sav1866 as a template because of its higher resolution (3.0 Å, against 3.7 Å for MsbA). This refined Sav1866-based homology model of CFTR has then been considered as the starting point for molecular dynamics simulations.
Model of the open form of the CFTR channel, generated by molecular dynamics
General features of the model
The initial structure of the present Sav1866-based model (left panel in Figs. 1, 2) is topologically very close to the template and to our previously published model [6]. This initial model, in an outward-facing conformation, possesses a channel-like structure, but this one is closed at the cytosolic side, with no possible entrance for ions from the cytoplasm. We next run molecular dynamics (MD) simulations in an appropriate environment for (1) exploring the short time scale conformational stability of the model, (2) refining it, and (3) possibly sampling other potential conformational states of the protein that lie near the starting point in the energy landscape.

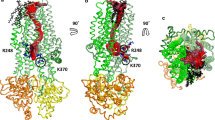
Comparison of the Sav1866-based CFTR model, before and after MD. A The MSD:NBD assembly. The CFTR model (encompassing aa 65–649 and 845–1,446) does not include the N-terminal, C-terminal and R regions, for which there is no template in the Sav1866 experimental 3D structure. The position of the lipid bilayer is symbolized in gray. The path of the polypeptidic chain can be followed with ribbons, colored according the architecture into domains (NBD1 light blue, NBD2 orange) or into blocks for the MSD (block 1 blue, block 2 green, block 3 yellow, block 4 red). ATP molecules are shown with solid spheres. At left the CFTR model before MD, closed at its cytosolic side (initial model). At the center full-open model of the CFTR channel (conformer 1). A ~15 Å upward movement of the region including F508 (shown in solid spheres) is observed, together with the creation of a large lateral tunnel, just under the membrane. A chloride ion is shown in green for illustration, at one of the entrances of this lateral tunnel, which is fully open and runs parallel to the membrane plane, from one side of the protein to the other (distance 47 Å). This lateral tunnel gives access to the main channel, which goes upward, perpendicularly to the membrane plane. At right closed form of the CFTR channel (closure at the extracellular part, conformer 2). B Ribbon representation of the MSD assembly. View of the same three conformers, which shows the four three-helix blocks of the MSDs from the extracellular side along the pseudo-binary axis and illustrates their displacement during MD. Positions 220 and 1,013 indicate the connections between blocks 1 and 2 and blocks 3 and 4, respectively. MSD1 runs from amino acid 65 to amino acid 381 and MSD2 from amino acid 845 to amino acid 1,177

The channel and its cytosolic access routes—global views. A Solid representation of the CFTR MSD:NBD assembly of the initial model, before MD (at left), conformer 1 (at the center, full-open channel) and conformer 2 (at right, closed channel). The main lateral tunnel (~47 Å long) is visible below the membrane, through its TM11/TM2 entrance in conformer 1. The position of the lipid bilayer is symbolized in gray. B Ribbon representations of the MSD assembly (four three-helix blocks) of the three same models, encircling the water-filled cavity constituting the ion channel (in solid, colored in blue green). The transition between the initial model (at left) and conformer 1 (at the center) gives rise to a well-defined channel and, importantly, to cytoplasmic lateral tunnels allowing access from the cytosol and merging with the channel. Note that the channels are arbitrarily cut at the extracellular sides, as they reach there the solvent. C Orthogonal view of the cytosolic end of the channel and of the lateral tunnels. These are labeled according to the TM helices participating in their formation. There are only two cytosolic entrances in conformer 2
To that aim, the locally minimized model of the 3D structure, with ATP molecules and magnesium ions in the two ATP-binding sites, was first embedded in a hydrated phospholipid bilayer, as described in details in the “Materials and methods” section. Then, the model was subjected to an unrestrained molecular dynamics (MD1) simulation at 310 K, over a period of 10 ns. We chose to work at 310 K (37 °C) as wild-type CFTR conductance is temperature sensitive and is enhanced at this physiological temperature compared to 28 °C [56]. The stability of the molecular assembly was monitored by following the internal energy of the protein and trend of the root-mean-square deviations (RMSD) of the Cα atoms positions as a function of time. These data, supporting the reliability of the simulation, are described in Online Resource 6(A) and 6(B). As in MD studies performed by another group on CFTR models, with similar relatively short simulation times [20, 21], the RMSD increased rapidly in the first nanosecond, before reaching equilibrium. This MD simulation was repeated three times (10 ns, each), to evaluate its robustness.
Analysis of the MD frames, in the different replica, showed that significant conformational changes rapidly occurred, resulting in a full-open channel, accessible from the cytosol. This transition is illustrated here with one particular frame of the first MD simulation (conformer 1, full-open channel, middle panel in Figs. 1, 2), reached as soon as 2.8 ns. Alternative rotamers have been considered in this conformer for a few amino acid side chains (the backbone being unchanged) that belong to the lateral tunnels, allowing an optimal description of these ones (see below).
We calculated RMSD between the initial model and the full-open channel model (conformer 1), to appreciate the structural differences between the canonical ABC exporter frame (that of the Sav1866 crystal structure, represented by the initial model) and the CFTR model at equilibrium in the open conformation. Mean RMSD, calculated over the whole MSD:NBD assembly, is moderately important [4.85 Å on 1187 Cα, Online Resource 6(C)] and is within the range of those observed for previous simulations [22, 23]. However, there were clear structural evolutions of specific domains/regions, as deduced from RMSD made after specific superimposition of those domains, although the global structure remained quite similar (Figs. 1, 2; Online Resource 7). We indeed observed a narrowing (~5 Å) and a twist at the level of the MSDs correlated with an expansion (also ~5 Å) at the level of the NBDs. The main changes observed indicate a movement of the transmembrane helices relative to each other (Figs. 1, 2; Online Resource 8). Contrasting with the RMSD for the whole MSD1:MSD2 assembly, much weaker RMSD values were observed when superimposing the structurally conserved elements of the MSDs described before (i.e., the blocks of three MSD consecutive helices TM1–3 (Block 1, highlighted in blue in Figs. 1, 2; Online Resource 8), TM4–6 (Block 2, green), TM7–9 (Block 3, yellow), and TM10–12 (Block 4, red) [Online Resource 6(C)]. Thus, the positions of the four three-helix blocks remarkably moved and twisted with respect to one another without drastically modifying their internal arrangement. The structural conservation of these elements is thus consistent with the invariance observed for these four three-helix blocks in the MSDs of crystallized ABC exporters, between outward and inward conformations (see before).
As reported previously in another MD study [21], the pore is characterized by a narrow bottleneck linking the outer vestibule to a large inner cavity, which, however, appeared larger than that observed in other MD studies (Fig. 3). These changes relative to the initial model are associated with the formation of a perfect salt bridge between R352 and D993, as experimentally observed by Cui et al. [5] (Figs. 3Bc, 5). Remarkably, in contrast to other MD simulations, we observed here lateral tunnels displayed within the ICLs, which allows access to the inner vestibule from the cytosol (Figs. 1, 2, 3Be, Bf). Also noteworthy is a shift (~15 Å) of the F508-containing helix within NBD1, associated with a correlated movement of ICL4 [Fig. 4; Online Resource 6(C)]. Although being a constant feature of MD1 and the three replica, which appeared once an energetic barrier has been overpassed, the amplitude of this shift is varying from one simulation to another one (Fig. 4).

The channel and its cytosolic access routes—cross-sections of the molecular surface of conformer 1. A Longitudinal cross-section of conformer 1 illustrating the global shape of the pore, with inner and outer vestibules, separated by a narrow constriction formed as TM6 (green) and TM12 (red) bring together. The inner vestibule is viewed at its narrowest point. The channel is open over its whole length, the crossing bridges observed at the level of R352 (salt bridge with D993) and M348 leading to its constriction. B Transversal views all along the channel, from the extracellular (a) to the intracellular (f) sides. The positions of these views, labeled a–f, are also shown in A

Movement of the F508 region. A Superimposition of the NBD1 α-subdomain (S489–D567) from the initial model, before MD (light blue and yellow) and conformer 1 (dark blue and orange). F508 is shown in yellow and orange, respectively. The RMSD between the Cα of the 63 amino acids shown in blue is 1.64 Å. B Focus on the F508 region, after a global superimposition of the CFTR 3D structure models (initial model, before MD—ICL4/ND1 interface colored yellow (labels in light brown); MD1 conformer 1—orange and a representative conformer of the MD1 replicas—pink). The nearly unchanged ICL1 is in blue at right. The MSDs + NBDs domains were considered for superimposition, except for RI (aa 403–436), ECL4 (aa 888–910) and LI (aa 1,178–1,202). The RMSD (1,186 Cα atoms) are 4.85 Å (initial model and MD1 replica) and 3.26 Å (conformer 1 and MD1 replica). A clear movement (~5 Å) of ICL4 upward and to the left occurred for the conformer 1/replica ensemble, associated with a large upward shift of the NBD1 500–515 region. This striking concerted movement, together with the establishment of the R353–D993 salt bridge might represent key markers for the activation of a functional channel, as illustrated here with the CFTR conformer 1
Conformer 1 remains essentially stable until the end of the simulation (10 ns), with only slow evolution [Online Resource 6(A)]. The same behavior is observed in the three replica of MD1. In particular, the shape of the pore appeared highly similar between the different simulations (Online Resource 9).
The channel and lateral tunnels
Analysis of the MD trajectories of the different replica showed that a ~5 Å upward movement of TM11 and of the two helices TM10 and TM12 (corresponding to block 4) and a tilting were observed during the very early steps. These were associated with a whole, concerted twist and displacement of MSD1 and MSD2 (Online Resource 8). In other words, there was a clear tilting, first of the fourth three-helix block (in red, TM10–TM12) and next of the three other three-helix blocks. These changes were associated with the establishment of an almost perfect salt bridge between R352 (TM6) and D993 (TM9), the distance between the side chain NH2 and O atoms decreasing from ~11 to 3 Å (Figs. 3Bc, 5). The existence of this salt bridge was reported to be a key marker of CFTR in its “full-open” conformation [5] and its establishment here thus supports the fact that this conformer obtained during MD (conformer 1) may represent a rather good approximation of the CFTR conformation in its “full-open” conformation. The movement of these two residues toward each other has also been reported in a previous MD simulation [20], leading to transient formation of a salt bridge (9 ns frame [21]). Here, the salt bridge appears to be quasi-permanent, once formed [see red line—52 value on the y axis in Online Resource 10(C)]. In addition, many other salt bridges between amino acids that are distant in the sequence (>7 aa) are also present in our model, at a level similar to that observed in the experimental 3D structure of Sav1866 (Online Resource 10).

The R352–D993 salt bridge. A longitudinal cross-section of the molecular surface of the CFTR conformer 1, perpendicularly to the channel axis highlights crossing bridges within the pore, leading to observe a narrowed “hexagonal” pore channel (involving TM12/11/2/1/3/6), as well as a smaller pentagonal duct (involving TM6/5/8/7/9). The two pores of the CFTR channel are filled, for illustration, with chloride ions (in green). Ten TM helices surround the channels, while TM6 is more largely participating in both pores than TM12. The two remaining helices (TM4 and TM10) line the secondary lateral tunnel (see Fig. 2)
The conformational rearrangement of the MSD clearly revealed the existence of a continuous channel able to carry ions (Cl−, I−), small charged compounds (e.g., Au(CN) −2 , HCO3 −, SCN−, glutathione) as well as molecules such as urea, from the cytosol toward the extracellular milieu. Green balls were used here in several figures for representing chloride ions within the channel. However, this has to be considered only for illustration purpose, to better visualize the conduction path through their volumes. Thus, these views do not take into account the repulsive effect between ions, which should be attenuated by water molecules, as well as do not attempt to define precise anion-binding sites. Remarkably, our model unambiguously revealed that the channel could be accessed via two cytoplasmic lateral tunnels, which run roughly parallel to the membrane plane and are located at the top of the ICLs, just beneath the lipid bilayer (Figs. 2, 3Be, Bf, 6). One of these lateral tunnels, which can be viewed as the main one, has a diameter similar to that of the main channel, whereas the second one, oriented at ~77° relatively to the former, is characterized by a more limited opening. Moreover, these two lateral tunnels and the lower part of the channel fuse together to constitute a large cavity (~15 × 6 Å), with a height perpendicular to the membrane plane of approximately 22 Å (Figs. 2, 3). This cavity likely corresponds to the widest part of the inner vestibule, which extends 14 Å below the membrane and 8 Å into the membrane space, up to the level of W356 (TM6) and S1149 (TM12), which face each other (Fig. 3; Online Resource 11). More upward, the channel is narrowed over a substantial part of its length (~14 Å), from R352 to I344 (TM6) and from N1148 to N1138 (TM12), due to a particular configuration of some side chains of TM6 and TM12, which fill the central space (Fig. 3; Online Resource 11). These side chains are those of the two amino acids (R352 and D993) forming the critical salt bridge, as well as those of M348 (TM6) and W1145 and S1141 (TM12). A clear narrowing in this region was also observed in other MD studies [21, 22]. As a consequence of the crossing bridges, we observed here that the central, narrow pore (called “hexagonal” path as it is lined by 6 helices: TM1, TM3, TM6, TM12, TM11, and TM2), is accompanied by a narrower, parallel duct (called “pentagonal” duct, as it is lined by 5 helices: TM5, TM8, TM7, TM9, and TM6). This secondary duct, which is separated from the main pore by the central bridge described above, has apparently not been observed in the other MD studies [21], principally as the movement of helices lining this pentagonal duct tends to occlude it. Closer to the top, the pore is widening over ~10 Å to form an almost rectangular architecture, with a narrowing (~20 × 6 Å) at the level of the upper side of the hydrophobic membrane, involving at its corners amino acids L100, I1131, T338, and V880, and at its center amino acids F337, G1130, and T1134. The amino acids that line this region are mainly hydrophobic (A96, P99, L100, F337, T338, S341, F342, V879, V880, I1131, T1134, and M1137) (Fig. 3; Online Resource 12). Noticeably, T and S might establish H-bonds with carbonyl atoms of the helices, therefore, masking their polarity. This rectangular part of the channel pore is constituted, for its two large sides, by the protein itself, and for its two small ones, by the hydrophobic parts of membrane lipids, over a length of ~8 Å (cross-sections 23–30, Online Resource 11). Finally, the size of the pore increases, leading to an extracellular vestibule of ~15 Å diameter, accessible from the extracellular space (Fig. 3; Online Resource 11).

Channel access from the cytosol. A The two entrances (9 by 5.5 Å) of the main lateral tunnel (entrances between TM5 and TM8, and between TM2 and TM11), passing through the protein via a linear path (length ~47 Å), from one side to the other. At left, a chloride ion is depicted in green and superimposed with a bicarbonate ion. Of note, the size of urea (CO(NH2)2) (not shown) is very similar to that of a bicarbonate ion. At the center is shown a molecule of glutathione. At right, a CFTR channel blocker (glibenclamide) is inserted, highlighting its widest part. B The two roughly circular entrances (diameter ~7 Å) of the secondary lateral tunnel, with a chloride ion (yellow) at left and right. The middle view shows the more constricted part of this lateral tunnel, at the level of two salt bridges linking R1048 to E1044 and to E979. The main and secondary lateral tunnels join the channel at the center, forming a large cavity, just below the cytoplasmic side of the membrane bilayer (see Fig. 2). C Up to three chloride ions have room to be theoretically inserted within the two entrances of the main lateral tunnel
The channel itself involves 10 of the 12 TM helices and the two remaining ones (TM4 and TM10) contribute to the formation of the secondary lateral tunnel (Figs. 2, 3). A hundred amino acids participate in the channel/tunnels structure (~40 for the main lateral tunnel, 25 for the secondary one and 45 for the channel itself). They are of miscellaneous nature (22 % basic residues, 24 % acidic or acidic-like residues, 16 % of P, G, T, or S and the remaining 38 % are hydrophobic) (see Table 1 for a list of essentially polar amino acids participating in the channel and lateral tunnels). The size of the channel itself is quite constant (surface between 50 and 90 Å2). The main lateral tunnel is larger than the secondary one (Fig. 6).
Of note, we observed in this CFTR conformer 1 a partial unwinding of the helical path over short portions of TM8, on one side of the lateral tunnel main entrance TM5–TM8 (H939–T943) and of TM9 (D979–D985) (an unwinding initiated in the Sav1866 between V128 and D133), as well as of TM2 (L145–I148), on the side of the other main entrance TM2–TM11. This unwinding at critical positions of the channel/tunnels ensemble is reminiscent of that recently observed in the P-glycoprotein homolog ABCB1, solved at high resolution (2.4 Å). Indeed, the ABCB1 TM4 segment, encompassing amino acids G277 to G286 and likely constituting one lateral border of the entrance for large hydrophobic molecules within the transmembrane region, displays partial unwinding and weak electron density. These features are suspected to allow the flexibility necessary for opening lateral tunnels.
The results of previous experimental studies analyzing the accessibility status of predicted pore-lining residues by mutagenesis and substituted cysteine accessibility mutagenesis (SCAM) are in good agreement with our present model of the full-open channel (conformer 1). This can be appreciated by analyzing Table 2, which reports the SCAM results obtained using both channel-permeant and channel-impermeant probes, as reported in [21]. These experimental results were compared to our predictions for TM6 and TM12, the two helices that are the major contributors to the anion conduction pore [1]. Logically, the periodicity of the helical path is well observed in the accessibility pattern observed either theoretically or experimentally, while most of amino acids within ECLs are accessible to solvent (e.g., the four contiguous positions L333, R334, K335, and I336, discussed in [57]—Table 2). Noteworthy is that, at the level of the central bridge within the channel, all the exposed residues that are reactive toward thiol-directed reagents when substituted with a cysteine are located within the “hexagonal” main pore, except one: T1142. R347 and T351 are indeed exposed within the secondary, “pentagonal” duct, but are not reactive to thiol-directed reagents (Online Resource 13). T1142, also exposed but reactive to thiol-directed reagents, is located at the very upper part of the pentagonal duct, suggesting that it may nonetheless may be accessible from the top of the pore, as S1141 (Online Resource 13). Altogether, these observations indicate that the residues from the “hexagonal” main pore are accessible to bulky cysteine-reactive reagents, while those of the secondary, “pentagonal” duct should not. This may suggest that the main “hexagonal” pore also constitutes the major way for ion conduction, while the “pentagonal” path should consist in a small bypass, or even as a buffer in the gating process, and therefore should play a less important role. This hypothesis is, however, difficult to test, as the impact of mutations in this region has generally minor effects (reviewed in [1]), except that of K95, which is located just up to the main hexagonal pore (Fig. 3Bb, see below). Noteworthy is that in the conformation adopted by conformer 1, the R347 side chain is free, and thus not forming a bridge with either D924 in TM8 [58] or D993 in TM9 [5]. This indicates that other conformations have probably to be sampled to understand the exact role of this amino acid in the CFTR gating behavior.
The major role played by the “hexagonal” main pore is furthermore supported by the fact that TM1 and TM11 (lining the “hexagonal” pore) have been ascribed as pore-lining based on functional and SCAM results [59–65], in contrast to the symmetric TM7 and TM5 [65], which line in the “pentagonal” duct. The particular, bridged architecture of the pore described here may thus particularly well account for the asymmetry observed for the CFTR channel, with a central main and narrowed pore lined by TM1, TM6, TM11, and TM12 [65] (Fig. 3Bc). Our model is also consistent with the fact that TM11 and TM6 appear to make similar but distinct contributions to the CFTR pore, as S1118 is less accessible than T338, according to the functional effects of their mutations [65] (Online Resource 14). Outside of TM6 and TM12, other predicted pore-lining residues, such as K95 (Fig. 3Bb), Q98 and P99 (TM1) [64] (upper part of the pore), L102 and R104 [66] (ECL1) (Online Resource 15) and R303 [65] (ICL2, inner vestibule, Fig. 3Bd) are also found accessible in the model obtained after MD. Finally, our model is also in good agreement with additional SCAM results [21] that suggested that TM3 and TM9 also contribute to the cytoplasmic portion of the pore: K190, D192, E193, G194, A196, L197, and F200 are indeed well exposed in the inner vestibule as are L989, P988, and L986 (Online Resource 16).
From Table 2, it can be noted that only a few positions disagree between our predictions and SCAM results. For instance, as in other MD simulations [21], there are several positions at the TM6 and TM12 N-termini (thus within ECL3 and ECL6, respectively) that are not reactive, although accessible on our model. Several hypotheses can be ruled out to explain such a discrepancy: a different local conformation might indeed exist due either to experimental artifact (possible effect of the substitution with a cysteine on the conformation) or modeling artifact (flexibility of some regions (loops) or conformations not explored by the MD simulation). We, however, note that SCAM results from independent studies, as well as mutagenesis studies, can sometimes be conflicting for a same position, possibly reflecting different experimental conditions. An example of such a disagreement is I1139 (TM12), which is reactive in the SCAM results reported by [21] but not in the SCAM results reported in two other, independent studies [66, 67]. The results of these last studies are thus in agreement with the buried position of this amino acid in our model (Table 2). Another example is T339 (TM6), which is reactive in the SCAM results reported by [21] but is not exposed in our model (Table 2; Online Resource 14). This buried position is, however, supported by the fact that several mutations of T339 resulted in a lack of functional channel expression and apparent misprocessing of the protein [68].
Finally, we note that 16 out of the 18 residues described in [25] as pore-lining residues, based on experimental data, are indeed exposed here to the aqueous conduction pore. This is significantly more than other previously published models of the open forms, including ours [6, 7, 21, 22, 25]. These 16 residues (K335, F337, T338, S341, I344, V345, M348, R352, T1134, N1138, S1141, T1142, Q1144, W1145, V1147, and N1148), have been discussed with respect to Table 2. Of note, four residues (out of those 16), which are located on TM12 (S1141, T1142, Q1144, and V1147), were initially (before MD) only poorly (or even not) accessible and became accessible during the MD-driven close-up of TM6 and TM12. The two residues still buried in our model are M1140 and V350. All the published models [6, 7, 21, 22, 25] agree with the buried position of M1140, whereas this residue could be modified by methanethiosulfonate (MTS) reagents, but only after channel activation [66]. The role of this specific residue in the pore awaits thus further investigations. A similar discrepancy exists for V350, which was reported by all the models as buried (Online Resource 17), but for which mutations have been associated with decreased sensitivity to open channel blockers [69]. However, in this last case, other experimental data confirmed the non-accessible character of this amino acid (Table 2, [21]). Furthermore, our structural analysis gave further predictive insights into the specific behavior of this region of TM6 (Online Resource 17).
The positively charged side chain of the lysine residue K95 occupies a central place in the channel, just above the “hexagonal” main pore, making it able to interact with permeant and blocker anions, i.e., inhibitors that act by directly blocking chloride movement through the open channel pore [70]. Accordingly, the charge of K95 is supposed to be involved in the attraction of the chloride ions into the pore, as its mutation causes outward-rectification of the current–voltage relationship [63] and dramatically reduce single channel conductance [62, 71]. Introduction of positive charges at positions that are here predicted to line the conduction path (amino acids belonging to the upper part of the pore (S341, I344, V355, and M348), lining the “hexagonal” pore (A349), or accessible on the upper part of the inner vestibule (R352 and Q353) (Table 2) indicated that the exact location of the charge is not crucial to support high conductance, although K95 plays a leading role [72]. Mutation of K95 also weakens the blocking effect of organic anions [63, 71], such as sulfonylureas (glibenclamide), which are able to reach the pore through the lateral main tunnel (Fig. 6), and then bind tightly at the level of K95 (Online Resource 18). Facing K95 are side chains of TM12 residues (e.g., M1140, V1147, and N1148) [69, 73], whose mutations also alter blockage by the glibenclamide and which may thus participate in its binding site. This one, also involving the stabilizing role of the W1145 indole ring, has been supported by a docking study performed on another CFTR model [22]. Other amino acids, such as R303 [74], have been reported to also participate in glibenclamide binding (reviewed in [70]). Binding to the site including R303 located within the inner vestibule, beneath the access to the “pentagonal” secondary duct, would block the passage of ions before the crossing bridge, while binding to K95 closes the upper part of the pore, just above the “hexagonal” main pore (Online Resource 18). The charged side chain of K978, located in ICL3, was also reported to be important for the mechanism of action of glibenclamide [75]. This one is located at the level of the floor of the inner vestibule, and thus participates in the main tunnel allowing access from the cytoplasm. K978 is thus likely playing a key role for the access of permeant and blocker anions to the pore, as other charged amino acids.
To that respect, it is noteworthy that the lateral tunnels concentrate the three-quarters of the basic amino acids, whereas the channel only contains one quarter (see Table 1 for the detailed composition of amino acids participating in the channel and lateral tunnels).
Some basic residues, such as K294, K946, K1165, and R1162 are located at the entrance of the tunnels, whereas other ones, such as R251, K190, K978, R1048, and R153 occupy inner positions (Fig. 3Bf). This distribution would ensure an efficient relay in the anion conduction path. Worth noting is that the very large organic anion suramin cannot be accommodated by the lateral tunnels due to its very large size. Accordingly, experimental data indicated that this compound occupies the entrance of the cytoplasmic pore, thus blocking the access for chloride ions [70, 74]. We observed that one of the sulfonate groups of the suramin head may firmly bind R303 through three salt bridges, while other interactions may also exist at the main tunnel entrance TM5/TM8 with R297 and K294 [Online Resource 19(A) and 19(B)]. This is again in agreement with experimental data, which demonstrated the critical role of R303 in binding suramin sulfonate groups [70, 74]. At the opposite entrance (TM2/TM11) of the same main tunnel, a similar situation may occur, with three salt bridges established with R153 and a H-bond with N1083 [Online Resource 19(C) and 19(D)].
Insights into the F508 region
Remarkably, in MD1, the global rearrangement of the whole set of transmembrane helices was accompanied by the shift of the NBD1 αα1 helix, at the extremity of which lies the residue F508 (Fig. 4a). The evolution of this region of the NBD1 α-subdomain appeared to be driven by the change of direction of the ICL4 coupling helix relative to the ICL1 one, from an initial configuration where they are parallel to each other (Fig. 4b). In the different replica, the amplitude of this global movement is variable, with intermediate states observed between the initial conformation and that adopted in conformer 1 (Fig. 4b). The magnitude of the shift of the NBD1 αα1 helix was actually the most important of the whole MSD:NBD assembly. Comparatively, P1306, the residue corresponding to F508 in NBD2, was only displaced by less than 4 Å (Online Resource 20). The regulatory insertion (RI), initially immersed in the solvent, appeared subjected to a more limited movement (max. 10 Å), stacking to NBD2 and to the non-hydrolysable ATP. The large extracellular loop ECL4 also showed only relatively limited movements. Altogether, these observations thus suggested that, relatively to the position of F508 before MD, this alternative position of F508 can only be reached if an energetic barrier of the energy landscape can be crossed and/or if a rearrangement of the MSD occurs toward a full-open channel conformation. This other conformational state of the protein, with an alternative position of F508, may constitute a key determinant for the functioning of wild-type CFTR.
After shift, the segment bearing F508 was still in contact with ICL4 but within a highly modified environment (Fig. 4). F508 is now in front of T1076 and L1077 (two amino acids belonging to the N-terminal extension of TM11, within ICL4), approximately 15 Å upward of its initial position. Moreover, a salt bridge between K1080 and E504 likely contributes to the stabilization of this assembly. F508 is now also in close proximity of Y1073 (Fig. 4). Of note, a perfect theoretical disulfide bridge in the double mutant F508C/Y1073C should be possible in this particular conformer [Online Resource 21 (B)]. The existence of the alternative position of F508 was further supported by the fact that the modification of F508C by benzyl-methanethiosulfonate (MTSBn), conserving the F508 aromatic character and restoring gating activity (lost for the F508C mutation in the open state-locked E1371S variant) [76], can be accommodated in both the initial and MD-generated models of CFTR [Online Resource 21 (A)].
The energy levels of the NBD1 α-subdomain (amino acids 489–567) calculated before and after shift were, respectively, −2,956 and −2,858 kJ/mol. In line with this, the energies of the concerned ICL4 segment (amino acids 1,053–1,180) were −726 and −871 kJ/mol, respectively, whereas they were, respectively, −3,892 and −3,863 kJ/mol, for the combined NBD1 + ICL4 segments. This suggested that the two positions have a similar stability. This was also in agreement with the very early onset of the F508 shift during MD and its subsequent stability during MD further supported this observation.
We further analyzed the sequence of the CFTR NBD1 α-subdomain, trying to better decipher the features that may underlie this conformational change. In ABC transporters, two short β-strands (β and β′) are present before the α-subdomain αα1 and αα2 helices, which can be viewed as the “arms of a nutcracker” and may play a critical role in signaling as they follow and precede the Q and X-loops, respectively [38] (Online Resource 1 and Online Resource 20). It is noteworthy here that, while present in CFTR NBD2, the small β–β′ β-sheet appears disorganized in CFTR NBD1. Indeed, the segment encompassing G500–T501–I502 appears to rotate and thus lose contact with its partner (I539–V540–L541), around a pivot constituted by P499 and G500 (Online Resource 20). Moreover, F508 is immediately followed by a glycine (G509), which at the difference of Y1307 (its corresponding residue in NBD2), cannot provide a side chain helping to stabilize the α-subdomain hydrophobic core. Altogether, all those specificities of NBD1 might account for the intrinsically less stable character of its α-subdomain.
Other regions
Finally, we also examined the evolution of other regions of the CFTR MSD:NBD assembly. Here, we first studied the four-helix bundle architecture, found at the basis of ICLs and possibly playing a role in the tight assembly of the CFTR cytoplasmic domains in the open form of the channel. We have indeed previously highlighted the functional importance of amino acids E267 (ICL2) and K1060 (ICL4), which are likely to form a salt bridge involved in the tight association of the ICLs [9]. When comparing our initial and full-open (conformer 1) models, we observed that the electrostatic interaction between E267 and K1060 remained conserved, although the four-helix bundle was slightly distorted (Online Resource 22).
Next, we studied two other regions that were not included in the Sav1866 MSD:NBD template, but correspond to short regions which were afterward modeled. First, the Regulatory Insertion (RI, from E403 to L435), between NBD1 strands β1 and ß2, was modeled similarly than previously reported [10]. During MD, the RI moved to come in a large direct contact with NBD2 and the non-hydrolysable ATP. Noticeably, three of the five phenylalanine groups of this long insertion interact with two phenylalanine groups of NBD2 [Online Resource 23(A)], and thereby constitute an aromatic cluster, which covers the ATP purine, opposite to W401. Such close contacts may reinforce the stability of ATP within the non-canonical ATP-binding site. One can also note that K413 establishes a salt bridge with E410 (E410 (3.1/4.2 Å) and contacts N416 (5.0 Å). Pseudo-symmetrically, K420 makes a salt bridge with D426 (2.9/3.8 Å) and contacts N417 (3.8 Å). In addition, the phosphorylable S422 is, in the conformer 1 structure, close to NBD2 K1334, the only basic amino acid in this region; the phosphate group and K1334 may thus form a salt bridge. Noticeably, a proline at this 422 position in chicken CFTR is thought to be associated with an increasing thermodynamic stability of the F508del protein [77]. Second, the Linker Insertion region (LI, from Y1182 to D1202) located upstream of NBD2 strand β1 is in a pseudo-symmetric position relatively to RI. It is shorter than RI and possesses, in its middle part, four successive hydrophobic amino acids (V1190 to I1193), which should favor the formation of a β-strand. In the hydrophobic cluster dictionary established on the basis of experimental 3D structures [78], a cluster formed by four consecutive hydrophobic amino acids (Binary code 1111, Peitsch code 15) is indeed associated at 86 % with extended (β-strand) structures. Taking into account the LI N- and C-terminal positions (defined by the Sav1866 template) and the neighboring NBD2 region, we hypothesized that a small β-sheet might be formed between Y1219–T1220 and the LI middle part (V1190 to I1193). This hypothesis was supported by MD [Online Resource Resource 23(B)], which showed that perfect N–H···O bonds linked E1221 and K1189, as well as M1191 and Y1219 (2.88 and 2.87 Å, respectively). Such a local structure is likely important for the functioning of CFTR, as it includes Y1219, the aromatic amino acid which stacks the purine cycle of the hydrolyzable ATP molecule in the canonical ATP-binding site. In this site, LI K1189 side chain also contacts the S962 (5.7 Å) located in the ICL3 coupling helix.
CF-causing missense mutations
Finally, we analyzed the full-open channel model of CFTR (conformer 1) in light of the data available on the “Clinical and Functional TRanslation of CFTR” (CFTR2) website (http://www.cftr2.org; [79]). The CFTR2 database provides information about how CFTR mutations affect the clinical outcome (genotype–phenotype correlation), the mutations being classified into the three following categories: CF-causing, of varying clinical consequence and non-CF-causing. Thus, we analyzed here a large series of specific mutations listed in the CFTR2 database and involving positions which can be mapped on our model of the open form of CFTR (amino acids 65–649 and 845–1,446). The model contains 43 out of the 46 missense mutations reported to be CF-causing, all the 11 missense mutations of varying clinical consequence, the two CF-causing deletions I507del and F508del, as well as 9 out of the 11 non-CF-causing mutations [Fig. 7; Online Resource 1 (positions underlined in green)]. Interestingly, the mutations studied are not distributed randomly, but they appear clustered around several hot spots.

CFTR2 missense mutations mapped onto the CFTR 3D model (full-open channel, conformer 1). A View from the top (extracellular side) of the mutations lying on the MSDs. B Salt bridges between the long ECLs: ECL1–ECL6 = D110–R1128 (4.0 and 5.0 Å) and R117–E1124 (5.5 and 5.8 Å); ECL3–ECL4 = R334–D891 (3.8 and 4.4 Å). The side chains of D891 and R1128 may adopt alternative positions and form a salt bridge, as this is possible in the closed form of the channel (conformer 2). Note that a salt bridge between ECL3 and ECL4, in a nearly symmetric position to R117–E1124, could not be present since there is no basic–acidic couple in this region. However, in addition to these salt bridges, Y109 (TM1) is H-bonded to to T1122 (TM11) and Y914 (TM8) is H-bonded to K929 (TM5), reinforcing the interactions between TMs. These contacts are nearly all conserved in conformer 2 (channel closed at the extracellular end), since the extracellular extremities of TM1–TM2 and TM11–TM12 move together (see Fig. 8). C Focus on the NBD1:ICL4 interface mainly lining the transition path of F508 from the initial model, before MD, to its final position in conformer 1 (see Fig. 4). D Focus on the NBD1:NBD2 interface, at the level of the canonical ATP-binding site
First, almost all CF-causing mutations involving residues located in the MSD transmembrane segments are encountered in MSD1 and generally concern positions lining the pore (G85E, E92K, D110H, P205S, R334W, I336K, T338I, S341P, R347H/R347P, and R352Q) (Fig. 7a). Such an asymmetry in natural CF-causing mutations is reminiscent of that observed for artificial mutations introduced into TM6 and TM12 with a view to test the accessibility of pore-lining residues (reviewed in [1], see above and Table 1). In addition, L206W and H199Y are situated nearby P205S, orientated toward the lipid bilayer. P67L lies in the loop between the N-terminal segment and the N-helix. R74W (which is reported to be of varying clinical consequence) is located in the vicinity of a large aromatic cluster, including F77, F78, W79, F81, F83, and Y84; the substitution of this arginine by a tryptophan might thus destabilize the local geometry. As already hinted to above, only two mutations are observed in MSD2, L927P, and M1101K, which both might disturb the conformation and behavior of the transmembrane helices within the lipid bilayer. Interestingly, amino acid R117, which is involved in the mutations R117C and R117H and is located in the first extracellular loop (ECL1) at the very beginning of TM2, can make a salt bridge with E1124 in ECL6 (distances of 5.5 and 5.8 Å) and might thus, among others, participate in the stabilization of the open form of the channel (Fig. 7b). The R117H mutation (varying clinical consequence) appears less severe than R117C (CF-causing), as histidine probably retains part of the attraction with the glutamate E1124 situated at 7.9 Å. Finally, two CF-causing mutations involve amino acids which are implied in salt bridges: (1) D110 in TM2 (mutation D110H; salt bridge with R1128, distances of 4.0 and 5.0 Å) and (2) R334 in ECL3 (mutation R334W; salt bridge with D891, distances of 3.8 and 4.4 Å) (Fig. 7b).
Second, CF-causing mutations in the ICLs involve residues located at the base of the four-helix bundle assembling the four internal ICL helices (symmetric positions in ICL1 (G178E G178R) and ICL3 (G970R), which cannot be substituted by any other amino acid because of steric hindrance reasons (Fig. 7c; Online Resource 2). S945L lies within the main lateral tunnel, and might thus impair access to the pore. Moreover, a large “hot spot” region for natural CFTR mutations is located at the NBD1:ICL4 interface, involving (1) six ICL4 positions (H1054D, G1061R, L1065P, R1066H/R1066C, F1074L, and L1077P), which line the path followed by F508 during the MD1 conformational transition from its initial to its final position, and (2) seven positions in NBD1 (S492F, I507del, F508del, V520F, A559T, R560K/R560T, and A561E) (Fig. 7c). Some other CFTR mutations of varying clinical consequences, such as F1052 V, G1069R, and R1070W/R1070Q complete the list in this region.
Third, at the level of the NBD1:NBD2 heterodimer, CF-causing mutations are concentrated within the canonical ATP-binding site (S549N, S549R, G551D/G551S, G1244E, S1251N, and S1255P) (Fig. 7d).
The effects of remaining mutations listed in CFTR2 database can also be well understood in light of our structural data: (1) A455E has indeed no room to be well adapted, (2) G1244E (mentioned above) also occurs in a well conserved position (position 23 in Online Resource 2), (3) L467P might disturb the helix in which it is included, (4) G1349D is in the non-canonical ATP-binding site and, alike its corresponding G551D, has no room to be well adapted in presence of ATP, and finally (5) N1303K might disturb the large Q-loop of the NBD2 α-helical subdomain (position 42 in Online Resource 1 and Online Resource 2).
Finally, the eight missense mutations reported as non-CF-causing, which can be examined in our model, are located in regions where there is large room, with no crucial neighbors. I1027T faces lipids, but its hydroxyl group likely hides its polarity by establishing a H-bond with the V1024 carbonyl group, within the same helix.
Of note, in contrast to the CF-causing missense mutations, the 43 mutations leading to a stop codon (out of a total of 45), which are all CF-causing, are almost uniformly distributed on the 3D model as this may be expected.
In conclusion, our MD-generated model (conformer 1) showed a continuous conduction pathway composed of a true well-formed channel. A crossing bridge forms a constriction over a limited part of its length. At the basis of the wide inner vestibule, two lateral tunnels, orthogonal to the channel, contain multiple positive charges and make possible the transfer of anions and small molecules from the cytosol toward the extracellular milieu. Our simulation also indicated the possibility of an alternate conformation of the F508 region, which might play an as yet unexplored role in CFTR inter-domain interaction and dynamics. This simulation has furthermore been enriched by the modeling of the regulatory insertion (within NBD1) and the linker insertion (between MSD2 and NBD2) and by reporting on the 3D model positions of mutations stored in the CFTR2 database. These appeared clustered in a few hot regions, within the pore and at the interfaces between domains.
A closed channel, obtained in two successive stages (conformer 2)
Then, we choose to reinitiate a new MD simulation (MD2) from this outstanding model of the full-open channel (conformer 1). Figure 8 illustrates the conformation changes at the level of the whole 3D structure along the MD frames, whereas Online Resource 24 shows the evolution of distances between F337, a key amino acid of TM6 and amino acids from other TM helices located at the same level. Conformer 1 remains nearly unchanged over 6.3 ns and then evolve toward a new conformer (intermediate conformer i) notably differing from the former by the concerted tilting of the upper (extracellular side) part of the TM1–TM2 and TM11–TM12 transmembrane helices of about 5 Å toward TM5 and TM7, which remained nearly fixed (Fig. 8c; Online Resource 24). This conformer i remained open at both sides of the channel and retained the alternative position of the F508 region unchanged, as observed in conformer 1 (Online Resource 25), as well as the R352 and D993 salt bridge. Then, after 18 ns from the beginning of MD2, a new stable conformer (conformer 2) appeared (Fig. 8d; Online Resource 24). Closure of the channel at its extracellular side was now observed, after the tilting of the upper parts of TM5–TM6 and TM7–TM8 by ~4 Å toward TM1 and TM12 (closed channel, right panel in Figs. 1 and 2). Again, the alternative position of the F508 region and the R352 and D993 salt bridge remained nearly unchanged. This conformer remained stable until the end of the simulation (30 ns). It is completely closed at the extracellular side, at the level of F337, but still partially open at the cytoplasmic side, through the main lateral tunnel entrance TM5/TM8 and the secondary one TM6/TM4 (the 2 other lateral tunnels entrances TM10/TM12 and TM2/TM11 are closed). Of note is the transient onset of conformer 2 within the conformer i region (Online Resource 24). Indeed, a conformer highly similar to conformer 2 (closed at the extracellular side) was observed at 10.89 ns during ~100 ps, preceded (at 10.23 ns) and followed (at 11.36 ns) by two intermediate conformers, with open channels at the extracellular side. Although this unique onset is not enough to be associated with a reversible event, it is tempting to hypothesize that this may corresponding to a flickering event, which may occur within burst of channel openings, but which make insignificant impact on overall chloride transport, in contrast to inter-burst closures. More work should, however, be performed to analyze further such a possibility.

Transition toward the closed form of the CFTR channel: comparison of the MSDs from the different conformers along the MD2 simulation. A Global lateral view. Conformers 1 (0 and 5 ns), conformers i (10 and 15 ns) and conformers 2 (20 and 30 ns). B Focus (lateral view) on the membrane portions of TM1–TM2 (blue), TM5–TM6 (green), TM7–TM8 (yellow), and TM11–TM12 (red). Dark colors are associated with conformers 1 (0 and 5 ns), whereas conformers i (10 and 15 ns) and conformers 2 (20 and 30 ns) are represented with light colors. C Extracellular view of the superimposition of conformer 1 (0 ns, dark colors) with conformer i (10 ns, light colors). TM1–TM2 and TM11–TM12 couples of helices move about 5/6 Å toward TM6 and TM7, while TM5–TM6 and TM7–TM8 are only slightly displaced. The channel, whose pore is shown, is still open. Of note is the onset during MD2 of a small β-sheet between successive hydrophobic amino acids (within the sequence) belonging to ECL3 (G330, I331, and I332) and ECL4 (A904, V905, and I906), which may reinforce the interaction between the four TM5–TM6 and TM7–TM8 helices. D A similar extracellular view of the superimposition of conformer 1 (0 ns, dark colors) with conformer 2 (30 ns, light colors). TM1–TM2 and TM11–TM12 couples of helices have nearly completed their movements toward TM6 and TM7, whereas TM5–TM6 and TM7–TM8 moved toward the center for about 3.7 Å, leading to the complete closure of the channel. The side chains of F337, I106, and L1133, taken as examples, are in close contact
The hypothesis that conformer 2 may correspond to a possible closed state of CFTR was further supported by comparison with recently published experimental 3D structures of ABC exporters obtained in inward-facing conformations. Indeed, we compared our models obtained by MD (conformers 1 and 2) to the experimental 3D structures of two orthologs of the heavy metal detoxification ABC exporter Atm1 [30, 31], both in inward-facing conformations and showing how glutathione binds to these proteins. Interestingly, the NBDs are only slightly separated, their two long symmetrical C-terminal helices interacting with each other, probably preventing for a part their large dissociation.
First, our full-open conformer of CFTR (conformer 1) shares with Atm1 (pdb 4mrs) the following features, highlighted after superimposition of the whole MSD1:MSD2 assembly (548 Cα atoms, RMSD 5.35 Å):
-
1.
A “lateral” tunnel in a similar position as the CFTR secondary tunnel, induced in the ABC exporters by the separation of NBDs (Online Resource 25),
-
2.
Similar pores within the channel, with only marked differences in the upper part, as Atm1 in its inward-facing conformation is closed at the extracellular side,
-
3.
Similar cavities at the level of the CFTR inner vestibule, occupied in Atm1 by the oxidized glutathione, which thus perhaps may also be accommodated by CFTR at a similar position (Online 27).
Second, the MSDs of our post-open, closed conformer of CFTR (conformer 2), shares with Atm1 the following striking features, in addition to those reported before and highlighted this time after superimposition of transmembrane segments of the MSD1:MSD2 assembly (114 Cα atoms, RMSD 2.55 Å):
-
1.
Similar 3D structures, although the initial CFTR model was build on the basis of an outward-facing conformation, and not on an inward one (as Atm1) (Fig. 9; Online Resource 28),
Fig. 9 
Comparison of the model of CFTR conformer 2 (closed channel, 30 ns) and the experimental 3D structure of N. aromaticivorans Atm1 (inward-facing conformation). Amino acids from the 12 transmembrane helices (TM) included in the membrane space were superimposed (114 Cα atoms, RMSD 2.55 Å). The coordinates of the N. aromaticivorans Atm1 were taken from pdb:4mrs (2.35 Å resolution). A Lateral views of the MSD1 block 2 (green) and MSD2 block 4 (red). CFTR and Atm1 ribbons are colored dark and light, respectively. Two amino acids that are very close to each other in TM6 and TM12 are shown at right (V302 (pink and green) for Atm1, F337 and G1130 for CFTR, also see Online Resource 29). B A view of the superimposition from the extracellular side, focusing at the level of the tight closure of the channel [as observed in Atm1 (pink and green) and predicted for CFTR (F337 and G1130)]
-
2.
Similar closures of the pore. This last feature is ensured in Atm1 by the two symmetric side chains of Val302 (V376 in S. cerevisiae Atm1), whereas the corresponding amino acids in CFTR are F337 (MSD1) and G1030 (MSD2). The superimposition leads to a quasi-perfect coincidence of these amino acids, as well as of the helices TM6 and TM12, to which they belong (Fig. 9). The structural importance of F337 is supported by experimental observations, indicating that this amino acid plays a key role in anion conductance and selectivity [80, 81]. F337 and its close neighbors ensure a perfect closure of the conformer 2 channel at its extracellular side (Online Resource 29).

In conclusion, this closed form of the CFTR channel (conformer 2) supports the hypothesis that channel closure occurred through relatively localized conformational changes on the upper part of the transmembrane domains, which are sufficient for closing an extracellular gate, in which F337 is a central player. This conformation, in which the NBDs are still associated, is very close to those observed within some experimental 3D structures of ABC exporters in inward-facing conformations, displaying limited separation of the NBDs.
Discussion
Although CFTR shares the common overall architecture of ABC exporters as well as some functional features, it is unique, primarily as it functions as an ATP-gated chloride channel. In ABC proteins, ATP-induced dimerization of the NBDs and subsequent hydrolysis-triggered domain separation are coupled to large movements of the transmembrane domains, leading to a wide opening toward the extracellular (outward-facing) or the intracellular (inward-facing) milieu, respectively. In this mechanism, also called the “switch model” [82], the two gates are alternatively closed to block the access to the corresponding side of the membrane. The situation is different for CFTR as it has been hypothesized that it evolved from a primordial ABC exporter by removing or atrophying its cytoplasmic gate [57, 67], behaving as a “degenerated transporter” or a “broken pump” [83–85]. The hypothesis of the lack of an internal gate in CFTR has been afterward supported at the experimental level, as residues deep inside the pore have been shown to be readily accessed by hydrophilic thiol reagents from the intracellular side of the membrane in both open and closed states [66, 67].
According to the analogy with ABC transporters, a channel open burst is initiated by the ATP-driven NBD1:NBD2 heterodimer formation and terminated by dimer dissociation [86]. However, because of the asymmetry at the ATP-binding sites [17, 18], there is now support for a model in which the catalytically inactive composite NBD1 (or site A) site remains closed throughout the gating cycle [34]. This mechanism, also referred to as the “constant contact model” [82], is now supported by several recent 3D structures of ABC exporters in inward-facing conformations, but with non-separated NBDs. The case of the heavy metal detoxification ABC exporter Atm1 [30, 31] has been discussed in details in the “Results” section of this article, but other recent structures now extend the reliability of such a model. Indeed, the recent 3D crystal structure and double electron resonance of TM287/TM288 in an inward-facing conformation highlights the quasi-invariance of contacts between the NBDs, even in the absence of nucleotides, the D-loop at the degenerated nucleotide-binding site linking the NBDs together [87]. On another hand, the 3D structure of an antibacterial peptide exporter (McjD) in a novel outward-occluded state [35] shares similarities with both inward-open MsbA and outward-open Sav1866, with the two NBDs are in the ATP-bound state.
The MD simulations we made here, using a refined model of the MSD:NBD assembly, well accounts for the specific properties of the CFTR channel versus ABC exporters. First, like for MD simulations performed by other groups, our model of the CFTR open channel (conformer 1) clearly shows the typical architecture defined from experimental data, as the outward-facing conformation deduced from the Sav1866 3D structure, used as template for modeling, fast evolved toward an architecture of an open channel with a wide inner vestibule, a narrow constriction and a smaller external vestibule (Fig. 3). This architecture is supported by various experimental data, including SCAM results provided by different groups, even though a few of them are conflicting, as well as binding sites for open channel blockers (see “Results”). Remarkably, as regards to other published MD simulations, our model of an open CFTR channel gives, at the atomic level, a structural view of a continuous and consistent path for ion conduction from the cytoplasm to the extracellular milieu. This path involves lateral tunnels that are displayed within the ICLs, which are described here (to our knowledge) for the first time at the atomic level on a structural point of view. Only Furukawa and colleagues [23] have mentioned a potential, but limited cytosolic portal, induced by the NBD dimerization, but which very likely differs from the one described here. The structural view presented here is in line with the previous observations of cytoplasmic portals based on electron microscopy (EM) [88] and functional data (ICL3 residues [89]). In particular, it was suggested from EM data that a cytoplasmic portal could be located at the level of TM11 (TM5) helices [88], a hypothesis consistent with our current model, in which these two helices participate in the main tunnel entrances (Fig. 2).
Thus, we highlight here that ICLs are important not only for contacting the NBDs but also for providing gateways to the conduction pore. Such lateral accesses to central pores have already been observed in other membrane systems, such as in the pore domain of the AcrB peristaltic pumps [90] or in ATP-gated channels of the P2X receptor family [91].
As described in the “Results” section, the lateral tunnels and the lower part of the channel (up to the two-third of the membrane bilayer) are abundantly lined by polar amino acids, including basic (R, K, H) and acidic (D, E) ones. These may ensure an efficient relay for anion conduction. More extensive experimental investigations are clearly needed to evaluate the importance of each of these amino acids at the floor of the inner vestibule and within the lateral tunnels. In the upper part of the channel (over a length of ~14 Å), this polar character gives way to mainly hydrophobic amino acids within the channel constriction. The extracellular vestibule is then again largely occupied by polar amino acids. This asymmetry along the path of conduction suggests that anions present in the cytosol are attracted by the numerous basic amino acids which line the lateral tunnels, finding a well polar environment before reaching a hydrophobic region in the last part of the transmembrane channel, which may accelerate their exit. Such an asymmetry along the conduction path is reminiscent of that described for a gated mechano-sensitive pentameric ion channel along its central pore [92].
Second, our relatively short MD simulations have also led to observe a closed conformation of the channel (conformer 2), in which ATP molecules are still associated with the NBDs, but perhaps more loosely at the level of the canonical binding site. Indeed, the general conformational change leading to the evolution of the pores and lateral tunnels of CFTR does not significantly modify the tight head-to-tail association of the NBDs, as assessed by the small variations of the ATP-binding site and of the experimentally observed bond between R555 (NBD1) and T1246 (NBD2) [93] (~2 Å for Cα between the initial model and conformer 2). These small variations are within the range suspected for this couple of amino acids to occur between open and closed channels [93]. However, as illustrated on Fig. 1, a continuous displacement of NBDs relatively to each other occurred between the different conformations. It is tempting to hypothesize that the conformation of the closed channel observed here (conformer 2) corresponds to the inter-burst C2 state of the linear three-state model [94, 95], interrupting channel openings (O) and contrasting with the long-closed state (C1), occurring following ATP hydrolysis and in which the NBD heterodimer could be completely dissociated. In the continuous presence of ATP, the channel should rarely visit this C1 state [95], which is not observed in the present modeling study.
This closed, “C2-like” conformation (conformer 2) essentially differs from the first full-open conformer by the compaction of the membraneous parts of TM1–TM2, TM11–TM12, TM5–TM6, and TM7–TM8, all the upper part of the helices being in close contact to each other and causing the closure of the channel on its extracellular side (Fig. 8; Online Resource 28). This result strongly supports the existence of an extracellular gate at the level of the critical amino acid F337. The limited movements allowing CFTR to jump from a full-open to a channel closed at the extracellular side (C2-like conformation) appear consistent with the high frequency of inter-burst closures during gating, the main part of the C2-like conformation remaining close to the full-open conformation (conformer 1). Remarkably, the transmembrane region of this closed channel conformer has a 3D structure very similar to the corresponding region of the N. aromaticivorans Atm1 exporter in an inward-facing conformation, solved at high resolution. This striking similarity provides strong support to our simulation and suggests that the different degrees of separation of the ICLs, and of the associated NBDs, do not significantly modify the final 3D state of such a channel closure. Indeed, between the CFTR conformer 2 and the inward-facing conformation of Atm1, the ICL2–ICL4 distance increases by approximately 20 Å.
Finally, we also compared our conformer 2 with our previous model of the closed form of the channel we built on the basis of the inward-facing, “closed-apo” conformer of V. cholerae MsbA (pdb 3b5x) [10] (Online Resource 30). Interestingly, this “closed-apo” model also approximates, as N. aromaticivorans Atm1, the CFTR conformer 2, but to a slightly lesser extent (RMSD 2.98 Å instead of 2.55 Å). In the three cases, helices TM1, TM6, TM7, and TM12 are nearly identically positioned around the pseudo-binary axis of the 3D structure. In conclusion, through MD simulation starting from an open form of the channel, we confirmed here the global architecture of a closed form that was anticipated by the recent 3D structures of ABC exporters in inward-facing conformations.
It remains still unclear, especially at the structural level, how the CFTR channel evolves during the gating cycle. From a simple situation, where it switches between “fully” open to “fully” closed state, more complexes scenario have been built, in which CFTR exhibits multiple states of sub-conductance in addition to a fully open state [5]. Two salt bridges, between R352 and D993 and between R347 and D924, have been proposed to contribute to the maintenance of the open pore architecture [58, 96]. Our MD simulation clearly had the effect of bringing forward the two helices TM6 and TM12, leading to the establishment of a critical salt bridge between R352 (TM6) and D993 (TM9), typical of the full-open channel conformation [5]. We did not observe here the D924–R347 or the triangular D924–R347–D993 salt bridges, which have been described as typical of early events in channel opening (s2 state [5]). In our model, at this level of the pore, TM9 is indeed separated from TM6 by TM5. Besides the fact that improving the quality of models in this region of high sequence divergence may still be possible, a plausible hypothesis is that the conformation specific of these early events has not been reached by the MD simulations presented here, and that more extensive exploration of the dynamic behavior of the CFTR channel is needed to further explore additional states. Of note is that in the initial model (before MD), the TM6 and TM8 helices are positioned in such a way that the salt bridge can be observed between R347 and D924 (Online Resource 31). This suggests that this particular topology should be considered in future investigations. It should also be noted that all the MD simulations were performed here without the presence of the R domain, for which no template still exists for classical homology modeling but which plays a critical role in the gating process [97]. Adding these critical regions in MD simulations should certainly give more insight into the subtle, different states of channel opening and gating mechanisms.
The R352–D993 salt bridge, together with amino acids in its vicinity (M348, W1145), locally constitutes a crossing bridge within the pore. The topological analysis we made here clearly indicates a protuberance of TM6 within the channel, consistent with its key role in conduction and the accessibility of cys-substituted residues to thiol reagents. In the full-open channel conformation generated by the MD simulation, TM6 has indeed ~¾ of its circumference exposed to the channel pore, subdivided for a part of its length into a narrowed main pore and a minor duct, in which TM6 participates. This structural feature particularly well accounts for the fact that six amino acids, belonging to two distinct thirds of the helix circumference (on the one hand I344, M348, and R352 and on the other hand S341, V345, and Q353) are all reactive to intracellularly applied probes when mutated in cysteine [57]. It has also been observed that channel gating is, however, affected through modification by positively charged MTSET of only one face, bearing I344, M348, and R352 [57], suggesting that TM6 undergoes helix rotation during gating of the CFTR pore. Examination of our model shows that I344, M348, and R352 are clearly orientated toward (or within) the “pentagonal” secondary duct, while S341, V345, and Q353 are at the end or within the “hexagonal” main pore. Thus, the observed structural dichotomy, correlating with functional properties, suggests that rotational movements of TM6, as proposed by [57], are not the only mechanism that can be proposed for switching between one state to another one. This hypothesis is supported by very recent data on the possible movements of TM helices during channel gating [98]. These experiments, gained using metal (Cd2+) bridges, shows that the key conformational changes that cause channel pore to open and close involve lateral separation and convergence of TM helices, rather than rotations and/or translations. Our models are in good agreement with these experimental data, as they shows that distances consistent with the formation of Cd2+ bridges are observed between TM6 and TM12 in the closed form of CFTR (and not necessarily in the open form—Online Resource 32).
Our modeling study also suggests that intrinsic flexibility within the membrane spanning domains is likely to play critical role in the channel function and gating mechanism. Indeed, the sequence alignment partly made with experimental 3D structures, which can be used as templates, clearly evidenced the presence of a π-bulge in CFTR TM6 (Online Resource 1), generated by a one-amino acid insertion relative to templates. π-bulges are often found in functional hot points of proteins [99, 100], and we hypothesized it is also the case here in the M348–P355 region of CFTR (also see Online Resource 17), as well as in TM2/TM8, in which such irregularities are also likely to occur. The importance of this position is further stressed out, as in TM6 of MRP4, the closest sequence neighbor of CFTR, phenylalanine F368, corresponding to the one-amino acid insertion (Q353 in CFTR), plays a crucial role in the substrate specific activity, as MRP4 W995 (aligned with CFTR W1145, which faces R352) and R998 [101]. Worth noting is that MRP4 and CFTR share a relatively high level of sequence identity (~36 % over the whole protein sequences, also including the N-terminal region, Online Resource 1), implying that their respective 3D structures are likely closely related, despite different functions (transport of large molecules and ion channel, respectively). This observation thus provides support to the fact that different functions can be ensured using a same 3D template.
Despite its variable amplitude in our MD simulations, the overall movement of the F508 region in NBD1 might possess structural as well as functional roles in the context of its interaction with other domains of CFTR. The functional role, possibly corresponding to the stabilization of an open conformer through the associated movement of ICL4, is especially supported by the results of an extensive mutagenesis study, which showed that while the aromatic side chain of F508 is not essential for the CFTR folding, it is important for the ion channel function [76]. This again emphasizes the importance of this region, which also plays a critical role in other ABC proteins, such as for yeast YOR1p [102] and human ABCG2, involved in urate transport and for which a F508del mimicking mutation impacts on its processing and stability [103]. Of note is that YOR1p, aligned here with the CFTR protein sequence (Online Resource 1) possesses as CFTR a long R region (51 aa), which moreover shares sequence similarities in the N-terminal part of the CFTR R domain [10]. Also of note is that the overall movement of the helix bearing F508 makes accessible the bi-phosphorylable SYDE motif located downstream of F508 [104].
In conclusion, the molecular dynamics simulation presented here illustrates, in a relatively short period of time, the rapid evolution of an initial, Sav1866-based model in an outward-facing conformation toward a full-open channel (conformer 1), stable for about 6 ns and then more slowly evolving toward an extracellular-closed channel (conformer 2). This transition takes place without clear separation of the NBDs, although numerous local movements are observed. These features are in agreement with the expected behavior of CFTR, for which opening and closing of the channel do not necessitate, in principle, large changes in the global 3D structure [1].
Thus, these models give new interesting insights into the MSD:NBD assembly behavior. However, an open question is still how the complex regulation of CFTR, mediated among others by the three other regions of CFTR (the N- and C-terminal extremities and the R domain), as well as by ATP hydrolysis, can be expressed in this context.
References
Linsdell P (2014) Functional architecture of the CFTR chloride channel. Mol Membr Biol 31:1–16
Wang Y, Wrennall J, Cai Z, Li H, Sheppard DN (2014) Understanding how cystic fibrosis mutations disrupt CFTR function: from single molecules to animal models. Int J Biochem Cell Biol 52:47–57
Rowe SM, Verkman AS (2013) Cystic fibrosis transmembrane regulator correctors and potentiators. Cold Spring Harb Perspect Med 3(7):a009761
Hollenstein K, Dawson JP, Locker KP (2007) Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol 17:412–418
Cui G, Freeman CS, Knotts T, Prince CZ, Kuang C, McCarty NA (2013) Two salts bridges differentially contribute to the maintenance of cystic fibrosis transmembrane conductance regulator (CFTR) channel function. J Biol Chem 288:20758–20767
Mornon J-P, Lehn P, Callebaut I (2008) Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci 65:2594–2612
Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV et al (2008) Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA 105:3256–3261
Dawson RJ, Locher KP (2006) Structure of a bacterial multidrug ABC transporter. Nature 443:180–185
Billet A, Mornon JP, Jollivet M, Lehn P, Callebaut I, Becq F (2013) CFTR: effect of ICL2 and ICL4 amino acids in close spatial proximity on the current properties of the channel. J Cyst Fibros 12:737–745
Mornon JP, Lehn P, Callebaut I (2009) Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci 66:3469–3486
Ward A, Reyes CL, Yu J, Roth CB, Chang G (2007) Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc Natl Aca Sci USA 104:19005–19010
Oldham ML, Davidson AL, Chen J (2008) Structural insights into ABC transporter mechanism. Curr Opin Struct Biol 18:726–733
Lukacs GL, Verkman AS (2012) CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med. 18:81–91
Du K, Lukacs GL (2009) Cooperative assembly and misfolding of CFTR domains in vivo. Mol Biol Cell 20:1903–1915
Du K, Sharma M, Lukacs GL (2005) The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol 12:17–25
Protasevich I, Yang Z, Wang C, Atwell S, Zhao X, Emtage S et al (2010) Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci 19:1917–1931
Wang C, Protasevich I, Yang Z, Seehausen D, Skalak T, Zhao X et al (2010) Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci 19:1932–1947
Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ et al (2012) Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 148:164–174
Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM et al (2012) Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148:150–163
Alexander C, Ivetac A, Liu X, Norimatsu Y, Serrano JR, Landstrom A et al (2009) Cystic fibrosis transmembrane conductance regulator: using differential reactivity toward channel-permeant and channel-impermeant thiol-reactive probes to test a molecular model for the pore. Biochemistry 48:10078–10088
Norimatsu Y, Ivetac A, Alexander C, Kirkham J, O’Donnell N, Dawson DC et al (2012) Cystic fibrosis transmembrane conductance regulator: a molecular model defines the architecture of the anion conduction path and locates a “bottleneck” in the pore. Biochemistry 51:2199–2212
Dalton J, Kalid O, Schushan M, Ben-Ta lN, Villà-Freixa J (2012) New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J Chem Inf Model 52:1842–1853
Furukawa-Hagiya T, Furuta T, Chiba S, Sohma Y, Sakurai M (2013) The power stroke driven by ATP binding in CFTR as studied by molecular dynamics simulations. J Phys Chem B 117:83–93
Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R et al (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323:1718–1722
Rahman K, Cui G, Harvey S, McCarty N (2013) Modeling the conformational changes underlying channel opening in CFTR. PLoS ONE 8:e74574
Ward AB, Szewczyk P, Grimard V, Lee CW, Martinez L, Doshi R et al (2013) Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci USA 110:13386–13391
Jin MS, Oldham ML, Zhang Q, Chen J (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490:566–569
Kodan A, Yamaguchi T, Nakatsu T, Sakiyama K, Hipolito C, Fujioka A et al (2014) Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc Natl Acad Sci USA 111:4049–4054
Shintre CA, Pike AC, Li Q, Kim JI, Barr AJ, Goubin S et al (2013) Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc Natl Acad Sci USA 110:9710–9715
Srinivasan V, Pierik AJ, Lill R (2014) Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 343:1137–1140
Lee JY, Yang JG, Zhitnitsky D, Lewinson O, Rees DC (2014) Structural basis for heavy metal detoxification by an Atm1-type ABC exporter. Science 343:1133–1136
Hohl M, Briand C, Grütter MG, Seeger MA (2012) Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat Struct Mol Biol 19:395–402
Basso C, Vergani P, Nairn AC, Gadsby DC (2003) Prolonged nonhydrolytic interaction of nucleotide with CFTR’s NH2-terminal nucleotide binding domain and its role in channel gating. J Gen Physiol 122:333–348
Szollosi A, Muallem DR, Csanády L, Vergani P (2011) Mutant cycles at CFTR’s non-canonical ATP-binding site support little interface separation during gating. J Gen Physiol 137:549–562
Choudhury HG, Tong Z, Mathavan I, Li Y, Iwata S, Zirah S et al (2014) Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc Natl Acad Sci USA 111:9145–9150
Callebaut I, Labesse G, Durand P, Poupon A, Canard L, Chomilier J et al (1997) Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell Mol Life Sci 53:621–645
Gaboriaud C, Bissery V, Benchetrit T, Mornon JP (1987) Hydrophobic cluster analysis: an efficient new way to compare and analyse amino acid sequences. FEBS Lett 224:149–155
Callebaut I, Eudes R, Mornon JP, Lehn P (2004) Nucleotide-binding domains of human cystic fibrosis transmembrane conductance regulator: detailed sequence analysis and three-dimensional modeling of the heterodimer. Cell Mol Life Sci 61:230–242
Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M et al (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23:282–293
Marti-Renom MA, Stuart A, Fiser A, Sánchez R, Melo F, Sali A (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291–325
Laskowski R, MacArthur M, Moss D, Thornton J (1993) PROCHECK—a program to check the stereochemical quality of protein structures. J App Cryst 26:283–291
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723
Pettersen E, Goddard T, Huang C, Couch G, Greenblatt D, Meng E et al (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Humphrey W, Dalke A, Schulten K (1996) VMD—visual molecular dynamics. J Mol Graph 14:33–38
Yu J, Zhou Y, Tanaka I, Yao M (2010) Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 26:46–52
Lomize M, Pogozheva I, Joo H, Mosberg H, Lomize A (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40:D370–D376
Aksimentiev A, Sotomayor M, Wells D (2012) Membrane proteins tutorial. http://www.ks.uiuc.edu/Training/Tutorials/science/membrane/mem-tutorial.pdf
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E et al (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
MacKerell AJ, Bashford D, Bellott M, Dunbrack RJ, Evanseck J, Field M et al (1998) All-hydrogen empirical potential for molecular modeling and dynamics studies of proteins using the CHARMM22 force field. J Phys Chem B 102:3586–3613
MacKerell AJ, Feig M, Brooks CI (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comp Chem 25:1400–1415
Feller S, MacKerell AJ (2000) An improved empirical potential energy function for molecular simulations of phospholipids. J Phys Chem B. 104:7510–7515
Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Ryckaert J-P, Ciccotti G, Berendsen H (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys 23:327–341
Darden T, York D, Pedersen L (1993) Particle mesh Ewald, an N·log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Feller S, Zhang Y, Pastor R, Brooks B (1995) Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys 103:4613–4621
Liu X, O’Donnell N, Landstrom A, Skach WR, Dawson DC (2012) Thermal instability of ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) channel function: protection by single suppressor mutations and inhibiting channel activity. Biochemistry 51:5113–5124
Bai Y, Li M, Hwang T (2010) Dual roles of the sixth transmembrane segment of the CFTR chloride channel in gating and permeation. J Gen Physiol 136:293–309
Cotten JF, Welsh MJ (1999) Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture of CFTR. Evidence for disruption of a salt bridge. J Biol Chem 274:5429–5435
Akabas MH, Kaufmann C, Cook TA, Archdeacon P (1994) Amino acid residues lining the chloride channel of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 269:14865–14868
Fatehi M, Linsdell P (2009) Novel residues lining the CFTR chloride channel pore identified by functional modification of introduced cysteines. J Membr Biol 228:151–164
Gao X, Bai Y, Hwang TC (2013) Cysteine scanning of CFTR’s first transmembrane segment reveals its plausible roles in gating and permeation. Biophys J 104:786–797
Ge N, Muise CN, Gong X, Linsdell P (2004) Direct comparison of the functional roles played by different transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem 279:55283–55289
Linsdell P (2005) Location of a common inhibitor binding site in the cytoplasmic vestibule of the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem 280:8945–8950
Wang W, El Hiani Y, Linsdell P (2011) Alignment of transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Gen Physiol 138:165–178
Wang W, El Hiani Y, Rubaiy HN, Linsdell P (2014) Relative contribution of different transmembrane segments to the CFTR chloride channel pore. Pflugers Arch 466:477–490
Quian F, El Hiani Y, Linsdell P (2011) Functional arrangement of the 12th transmembrane region in the CFTR chloride channel pore based on functional investigation of a cysteine-less CFTR variant. Eur J Physiol. 462:559–571
Bai Y, Li M, Hwang T (2011) Structural basis for the channel function of a degraded ABC transporter, CFTR (ABCC7). J Gen Physiol 138:495–507
Linsdell P, Zheng SX, Hanrahan JW (1998) Non-pore lining amino acid side chains influence anion selectivity of the human CFTR Cl-channel expressed in mammalian cell lines. J Physiol 512:1–16
Cui G, Song B, Turki HW, McCarty NA (2012) Differential contribution of TM6 and TM12 to the pore of CFTR identified by three sulfonylurea-based blockers. Pflugers Arch 463:405–418
Linsdell P (2014) Cystic fibrosis transmembrane conductance regulator chloride channel blockers: pharmacological, biophysical and physiological relevance. World J Biol Chem 5:26–39
Zhou JJ, Li MS, Qi J, Linsdell P (2010) Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J Gen Physiol 135:229–245
El Hiani Y, Linsdell P (2012) Tuning of CFTR chloride channel function by location of positive charges within the pore. Biophys J 103:1719–1726
Gupta J, Lindsell P (2003) Extent of the selectivity filter conferred by the sixth transmembrane region in the CFTR chloride channel pore. Mol Membr Biol 20:45–52
St Aubin CN, Zhou JJ, Linsdell P (2007) Identification of a second blocker binding site at the cytoplasmic mouth of the cystic fibrosis transmembrane conductance regulator chloride channel pore. Mol Pharmacol 71:1360–1368
Melin P, Hosy E, Vivaudou M, Becq F (2007) CFTR inhibition by glibenclamide requires a positive charge in cytoplasmic loop three. Biochim Biophys Acta 1768:2438–2446
Cui L, Aleksandrov L, Hou YX, Gentzsch M, Chen JH, Riordan JR et al (2006) The role of cystic fibrosis transmembrane conductance regulator phenylalanine 508 side chain in ion channel gating. J Physiol 572:347–358
Aleksandrov A, Kota P, Cui L, Jensen T, Alekseev A, Reyes S et al (2012) Allosteric modulation balances thermodynamic stability and restores function of ΔF508 CFTR. J Mol Biol 419:41–60
Eudes R, Le Tuan K, Delettré J, Mornon JP, Callebaut I (2007) A generalized analysis of hydrophobic and loop clusters within globular protein sequences. BMC Struct Biol 7:2
Sosnay P, Siklosi K, Van Goor F, Kaniecki K, Yu H, Sharma N et al (2013) Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 45:1160–1167
Linsdell P (2001) Relationship between anion binding and anion permeability revealed by mutagenesis within the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Physiol 531:51–66
Linsdell P, Evagelidis A, Hanrahan JW (2000) Molecular determinants of anion selectivity in the cystic fibrosis transmembrane conductance regulator chloride channel pore. Biophys J 78:2973–2982
George A, Jones P (2012) Perspectives on the structure–function of ABC transporters: the switch and constant contact models. Prog Biophys Mol Biol 109:95–107
Gadsby DC (2009) Ion channels versus ion pumps: the principal difference, in principle. Nat Rev Mol Cell Biol 10:344–352
Chen T, Hwang T (2008) CLC-0 and CFTR: chloride channels evolved from transporters. Physiol Rev 88:351–387
Miller C (2010) CFTR: break a pump, make a channel. Proc Natl Acad Sci USA 107:959–960
Verdon G, Albers SV, Dijkstra BW, Driessen AJ, Thunnissen AM (2003) Crystal structures of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: nucleotide-free and nucleotide-bound conformations. J Mol Biol 330:343–358
Hohl M, Hürlimann LM, Böhm S, Schöppe J, Grütter MG, Bordignon E et al (2014) Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporter. Proc Natl Acad Sci USA 111:11025–11030
Rosenberg MF, O’Ryan LP, Hughes G, Zhao Z, Aleksandrov LA, Riordan JR et al (2011) The cystic fibrosis transmembrane conductance regulator (CFTR): three-dimensional structure and localization of a channel gate. J Biol Chem 286:42647–42654
El Hiani Y, Linsdell P (2012) Role of the juxtamembrane region of cytoplasmic loop 3 in the gating and conductance of the cystic fibrosis transmembrane conductance regulator chloride channel. Biochemistry 51:3971–3981
Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM (2006) Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313:1295–1298
Samways D, Khakh B, Dutertre S, Egan T (2011) Preferential use of unobstructed lateral portals as the access route to the pore of human ATP-gated ion channels (P2X receptors). Proc Natl Acad Sci USA 108:13800–13805
Chang G, Spencer RH, Lee A, Barclay MT, Rees DC (1996) Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science 282:2220–2226
Vergani P, Lockless SW, Nairn AC, Gadsby DC (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433:876–880
Winter MC, Sheppard DN, Carson MR, Welsh MJ (1994) Effetc of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophys J 66:1398–1403
Jih K, Hwang T (2012) Nonequilibrium gating of CFTR on an equilibrium theme. Physiology 27:351–361
Cui G, Zhang ZR, O’Brien AR, Song B, McCarty NA (2008) Mutation at arginine 352 alters the pore architecture of CFTR. J Membr Biol 222:91–106
Wang W, Wu J, Bernard K, Li G, Wang G, Bevensee M et al (2010) ATP-independent CFTR channel gating and allosteric modulation by phosphorylation. Proc Natl Acad Sci USA 107:3888–3893
El Hiani Y, Linsdell P (2014) Metal bridges illuminate transmembrane domain movements during gating of the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem (in press)
Cooley R, Arp D, Karplus P (2010) Evolutionary origin of secondary structures: π-helices as cryptic but widespread insertional variations of α-helices that enhance protein functionality. J Mol Biol 404:232–246
Riek R, Graham R (2011) The elusive π-helix. J Struct Biol 173:153–160
Wittgen H, van den Heuvel J, Krieger E, Schaftenaar G, Russel F, Koenderink J (2012) Phenylalanine 368 of multidrug resistance-associated protein 4 (MRP4/ABCC4) plays a crucial role in substrate-specific transport activity. Biochem Pharmacol 84:366–373
Pagant S, Halliday JJ, Kougentakis C, Miller EA (2010) Intragenic suppressing mutations correct the folding and intracellular traffic of misfolded mutants of Yor1p, a eukaryotic drug transporter. J Biol Chem 285:36304–36314
Sarankó H, Tordai H, Telbisz Á, Özvegy-Laczka C, Erdős G, Sarkadi B et al (2013) Effects of the gout-causing Q141K polymorphism and a CFTR ΔF508 mimicking mutation on the processing and stability of the ABCG2 protein. Biochem Biophys Res Commun 437:140–145
Venerando A, Cesaro L, Marin O, Donella-Deana A, Pinna LA (2014) A “SYDE” effect of hierarchical phosphorylation: possible relevance to the cystic fibrosis basic defect. Cell Mol Life Sci 71:2193–2196
Acknowledgments
This work is supported by grants of the French association Vaincre La Mucoviscidose, and computing facilities provided by IDRIS (projects X2013072174 and X2014077206) and Qarnot Computing. We thank Benjamin Boucherle, Romain Haudecoeur, Antoine Fortuné, and Jean-Luc Décout (UMR5063 UJF/CNRS, Université Joseph Fourier, Grenoble) as well as Clément Boinot, Mathilde Jollivet, and Frédéric Becq (FRE3511, CNRS, Université de Poitiers) for fruitful discussions. The indirect contribution of K. Locher and co-workers, by solving several years ago the X-ray structure of Sav1866, which made this study possible, was also appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
J.-P. Mornon and B. Hoffmann contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mornon, JP., Hoffmann, B., Jonic, S. et al. Full-open and closed CFTR channels, with lateral tunnels from the cytoplasm and an alternative position of the F508 region, as revealed by molecular dynamics. Cell. Mol. Life Sci. 72, 1377–1403 (2015). https://doi.org/10.1007/s00018-014-1749-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1749-2