
Abstract
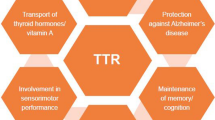
Transthyretin (TTR) (formerly, thyroxine binding prealbumin) is an evolutionarily conserved serum and cerebrospinal fluid protein that transports holo-retinol-binding protein and thyroxine. Its serum concentration has been widely used to assess clinical nutritional status. It is also well known that wild-type transthyretin and approximately 100 different mutants give rise to a variety of forms of systemic amyloid deposition. It has been suspected and recently established that TTR can suppress the Alzheimer’s disease phenotype in transgenic animal models of cerebral Aβ deposition. Thus, while TTR is a systemic amyloid precursor, in the brain it seems to have an anti-amyloidogenic effect. TTR is found in other organs as a result of local synthesis or transport, suggesting that it may have other, as yet undiscovered, functions. It is possible that its capacity to bind many classes of compounds allows it to serve as an endogenous detoxifier of molecules with potential pathologic effects.
Similar content being viewed by others
References
Cavallaro T, Martone RL, Dwork AJ, Schon EA, Herbert J (1990) The retinal pigment epithelium is the unique site of transthyretin synthesis in the rat eye. Invest Ophthalmol Vis Sci 31:497–501
Herbert J, Wilcox JN, Pham KC, Fremeau RT, Zaviani M, Dwork A, Soprano D, Makover A, Goodman DS, Zimmerman EZ, Roberts JL, Schon EA (1986) Transthyretin: a choroid plexus-specific transport protein in human brain. Neurology 36:900–911
Jacobsson B, Collins VP, Grimelius L, Pettersson T, Sandstedt B, Carlstrom A (1989) Transthyretin immunoreactivity in human and porcine liver, choroid plexus, and pancreatic islets. J Histochem Cytochem 37:31–37
Goodman DS (1986) Statement regarding nomenclature for the protein known as prealbumin, which is also (recently) called transthyretin. In: Glenner GG, Osserman EF, Benditt EP, Calkins E, Cohen AS, Zucker-Franklin D (eds) Amyloidosis. Plenum, New York, pp 287–288
Ingenbleek Y, Young VR (2002) Significance of transthyretin in protein metabolism. Clin Chem Lab Med 40:1281–1291
Potter MA, Luxton G (2002) Transthyretin measurement as a screening tool for protein calorie malnutrition in emergency hospital admissions. Clin Chem Lab Med 40:1349–1354
Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD (2007) A primer of amyloid nomenclature. Amyloid 14:179–183
Pitkanen P, Westermark P, Cornwell GGIII (1984) Senile systemic amyloidosis. Am J Pathol 117:391–399
Kyle RA, Gertz MA, Linke RP (1992) Amyloid localized to tenosynovium at carpal tunnel release. Immunohistochemical identification of amyloid type. Am J Clin Pathol 97:250–253
Rocken C, Saeger W, Linke RP (1994) Gastrointestinal amyloid deposits in old age. Pathol Res Pract 190:641–649
Buxbaum JN (2007) Transthyretin and the transthyretin amyloidoses. In: Uversky VN, Fink A (eds) Protein misfolding, aggregation, and conformational diseases. Springer, Santa Cruz, pp 259–283
Jacobson DR, McFarlin DE, Kane I, Buxbaum JN (1992) Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet 89:353–356
Hurshman AR, White JT, Powers ET, Kelly JW (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43:7365–7381
Teng MH, Yin JY, Vidal R, Ghiso J, Kumar A, Rabenou R, Shah A, Jacobson DR, Tagoe C, Gallo G, Buxbaum J (2001) Amyloid and nonfibrillar deposits in mice transgenic for wild-type human transthyretin: a possible model for senile systemic amyloidosis. Lab Invest 81:385–396
Sousa MM, Cardoso I, Fernandes R, Guimaraes A, Saraiva MJ (2001) Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol 159:1993–2000
Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN (2004) Tissue damage in the amyloidoses: transthyretin monomers and non-native oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA 101:2817–2822
Andersson K, Olofsson A, Nielsen EH, Svehag SE, Lundgren E (2002) Only amyloidogenic intermediates of transthyretin induce apoptosis. Biochem Biophys Res Commun 294:309–314
Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T (2008) Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci USA 105:2681–2686
Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ (2007) Accelerated Abeta deposition in APPswe/PS1deltaE9 mice with hemizygous deletions of TTR (transthyretin). J Neurosci 27:7006–7010
Sousa JC, Grandela C, Fernandez-Ruiz J, de Miguel R, de Sousa L, Magalhaes AI, Saraiva MJ, Sousa N, Palha JA (2004) Transthyretin is involved in depression-like behaviour and exploratory activity. J Neurochem 88:1052–1058
Shirahama T, Skinner M, Westermark P, Rubinow A, Cohen AS, Brun A, Kemper TL (1982) Senile cerebral amyloid. Prealbumin as a common constituent in the neuritic plaque, in the neurofibrillary tangle, and in the microangiopathic lesion. Am J Pathol 107:41–50
Schwarzman AL, Tsiper M, Wente H, Wang A, Vitek MP, Vasiliev V, Goldgaber D (2004) Amyloidogenic and anti-amyloidogenic properties of recombinant transthyretin variants. Amyloid 11:1–9
Schwarzman AL, Goldgaber D (1996) Interaction of transthyretin with amyloid beta-protein: binding and inhibition of amyloid formation. In: Bock GR, Goode JA (eds) The nature and origin of amyloid fibrils. Wiley, New York, pp 146–164
Link CD (1995) Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA 92:9368–9372
Stein TD, Johnson JA (2002) Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci 22:7380–7388
Sousa JC, Cardoso I, Marques F, Saraiva MJ, Palha JA (2006) Transthyretin and Alzheimer’s disease: where in the brain? Neurobiol Aging 28:713–718
Hovatta I, Schadt EE, Libiger O, Schork NJ, Lockhart DJ, Barlow C, Zapala MA, Broide RS (2007) DNA variation and brain region-specific expression profiles exhibit different relationships between inbred mouse strains: implications for eQTL mapping studies. Genome Biol 8:R25
Sousa JC, Marques F, Dias-Ferreira E, Cerqueira JJ, Sousa N, Palha JA (2007) Transthyretin influences spatial reference memory. Neurobiol Learn Mem 88:381–385
Fleming CE, Saraiva MJ, Sousa MM (2007) Transthyretin enhances nerve regeneration. J Neurochem 103:831–839
Richardson SJ, Lemkine GF, Alfama G, Hassani Z, Demeneix BA (2007) Cell division and apoptosis in the adult neural stem cell niche are differentially affected in transthyretin null mice. Neurosci Lett 421:234–238
Brouillette J, Quirion R (2008) Transthyretin: a key gene involved in the maintenance of memory capacities during aging. Neurobiol Aging 29:1721–1732
Serot JM (1997) Cerebrospinal fluid transthyretin: aging and late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry 63:506–508
Hansson SF, Andreasson U, Wall M, Skoog I, Andreasen N, Wallin A, Zetterberg H, Blennow K (2009) Reduced levels of amyloid-beta-binding proteins in cerebrospinal fluid from Alzheimer’s disease patients. J Alzheimers Dis 16:389–397
Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH, Sousa N, Correia-Neves M, Palha JA (2009) Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab 29:921–932
Liu L, Murphy RM (2006) Kinetics of inhibition of beta-amyloid aggregation by transthyretin. Biochemistry 45:15702–15709
Schreiber G, Richardson SJ (1997) The evolution of gene expression, structure and function of transthyretin. Comp Biochem Physiol 116B:137–160
Shoji S, Nakagawa S (1988) Serum prealbumin and retinol-binding protein concentrations in Japanese-type familial amyloid polyneuropathy. Eur Neurol 28:191–193
Filteau SM (2000) Use of the retinol-binding protein: transthyretin ratio for assessment of vitamin A status during the acute-phase response. Br J Nutr 83:513–520
Baures PW, Peterson SA, Kelly JW (1998) Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem 6:1389–1401
Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW (2004) Diflunisal analogues stabilize the native state of transthyretin Potent inhibition of amyloidogenesis. J Med Chem 47:355–374
Johnson SM, Connelly S, Wilson IA, Kelly JW (2009) Toward optimization of the second aryl substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem 52:1115–1125
Johnson SM, Connelly S, Wilson IA, Kelly JW (2008) Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J Med Chem 51:6348–6358
Johnson SM, Connelly S, Wilson IA, Kelly JW (2008) Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J Med Chem 51:260–270
Reixach N, Adamski-Werner SL, Kelly JW, Koziol J, Buxbaum JN (2006) Cell based screening of inhibitors of transthyretin aggregation. Biochem Biophys Res Commun 348:889–897
White JT, Kelly JW (2001) Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc Natl Acad Sci USA 98:13019–13024
Roche M, Rondeau P, Singh NR, Tarnus E, Bourdon E (2008) The antioxidant properties of serum albumin. FEBS Lett 582:1783–1787
Varshney A, Sen P, Ahmad E, Rehan M, Subbarao N, Khan RH (2009) Ligand binding strategies of human serum albumin: how can the cargo be utilized? Chirality. doi:10.1002/chir.20709
Steinrauf LK, Cao Y, Hamilton J, Murrell J, Liepnieks JJ, Benson MD (1991) Preparation and crystallization of human transthyretin (prealbumin) variants. Biochem Biophys Res Commun 179:804–809
Stockigt JR, Lim CF, Barlow JW, Wynne KN, Mohr VS, Topliss DJ, Hamblin PS, Sabto J (1985) Interaction of furosemide with serum thyroxine-binding sites: in vivo and in vitro studies and comparison with other inhibitors. J Clin Endocrinol Metab 60:1025–1031
Larsen PR (1972) Salicylate-induced increases in free triiodothyronine in human serum. Evidence of inhibition of triiodothyronine binding to thyroxine-binding globulin and thyroxine-binding prealbumin. J Clin Invest 51:1125–1134
Pfeffer BA, Becerra SP, Borst DE, Wong P (2004) Expression of transthyretin and retinol binding protein mRNAs and secretion of transthyretin by cultured monkey retinal pigment epithelium. Mol Vis 10:23–30
Kawaguchi R, Yu J, Honda J, Hu J, Whitelegge J, Ping P, Wiita P, Bok D, Sun H (2007) A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 315:820–825
Gaur VP, De Leeuw AM, Milam AH, Saari JC (1990) Localization of cellular retinoic acid-binding protein to amacrine cells of rat retina. Exp Eye Res 50:505–511
Ong DE, Davis JT, O’Day WT, Bok D (1994) Synthesis and secretion of retinol-binding protein and transthyretin by cultured retinal pigment epithelium. Biochemistry 33:1835–1842
Barouch FC, Benson MD, Mukai S (2004) Isolated vitreoretinal amyloidosis in the absence of transthyretin mutations. Arch Ophthalmol 122:123–125
Ando Y, Terazaki H, Nakamura M, Ando E, Haraoka K, Yamashita T, Ueda M, Okabe H, Sasaki Y, Tanihara H, Uchino M, Inomata Y (2004) A different amyloid formation mechanism: de novo oculoleptomeningeal amyloid deposits after liver transplantation. Transplantation 77:345–349
Suk JY, Zhang F, Balch WE, Linhardt RJ, Kelly JW (2006) Heparin accelerates gelsolin amyloidogenesis. Biochemistry 45:2234–2242
Mullins RF, Russell SR, Anderson DH, Hageman GS (2000) Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 14:835–846
Cras-Meneur C, Inoue H, Zhou Y, Ohsugi M, Bernal-Mizrachi E, Pape D, Clifton SW, Permutt MA (2004) An expression profile of human pancreatic islet mRNAs by serial analysis of gene expression (SAGE). Diabetologia 47:284–299
Westermark GT, Westermark P (2008) Transthyretin and amyloid in the islets of Langerhans in type-2 diabetes. Exp Diabetes Res 2008: Article ID 429274
Ando Y, Yi S, Nakagawa T, Ikegawa S, Hirota M, Miyazaki A, Araki S (1991) Disturbed metabolism of glucose and related hormones in familial amyloidotic polyneuropathy: hypersensitivities of the autonomic nervous system and therapeutic prevention. J Auton Nerv Syst 35:63–70
Nagasaka T, Togashi S, Watanabe H, Iida H, Nagasaka K, Nakamura Y, Miwa M, Kobayashi F, Shindo K, Shiozawa Z (2009) Clinical and histopathological features of progressive-type familial amyloidotic polyneuropathy with TTR Lys54. J Neurol Sci 276:88–94
Itoh N, Hanafusa T, Miyagawa J, Tamura S, Inada M, Kawata S, Kono N, Tarui S (1992) Transthyretin (prealbumin) in the pancreas and sera of newly diagnosed type I (insulin-dependent) diabetic patients. J Clin Endocrinol Metab 74:1372–1377
Refai E, Dekki N, Yang SN, Imreh G, Cabrera O, Yu L, Yang G, Norgren S, Rossner SM, Inverardi L, Ricordi C, Olivecrona G, Andersson M, Jornvall H, Berggren PO, Juntti-Berggren L (2005) Transthyretin constitutes a functional component in pancreatic beta-cell stimulus-secretion coupling. Proc Natl Acad Sci USA 102:17020–17025
Dowling P, Shields W, Rani S, Meleady P, Henry M, Jeppesen P, O’Driscoll L, Clynes M (2008) Proteomic analysis of conditioned media from glucose responsive and glucose non-responsive phenotypes reveals a panel of secreted proteins associated with beta cell dysfunction. Electrophoresis 29:4141–4149
Sundsten T (2006) The use of proteomics in identifying differentially expressed serum proteins in humans with type 2 diabetes. Proteome Sci 4:22. doi:10.1186/1477-5956-4-22
Sundsten T, Ostenson CG, Bergsten P (2008) Serum protein patterns in newly diagnosed type 2 diabetes mellitus—influence of diabetic environment and family history of diabetes. Diabetes Metab Res Rev 24:148–154
Kisilevsky R (2000) The relation of proteoglycans, serum amyloid P and apo E to amyloidosis current status, 2000. Amyloid 7:23–25
Kisilevsky R, Szarek WA, Ancsin J, Vohra R, Li Z, Marone S (2004) Novel glycosaminoglycan precursors as antiamyloid agents: part IV. J Mol Neurosci 24:167–172
Acknowledgements
This work was supported by National Institute on Aging Grants R01 AG30027 (to J.N.B.) and R01 AG19259 (to J.N.B.), the W. M. Keck Foundation (to J.N.B.) and The American Heart Association BGIA award 0865061F (to N.R.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Buxbaum, J.N., Reixach, N. Transthyretin: the servant of many masters. Cell. Mol. Life Sci. 66, 3095–3101 (2009). https://doi.org/10.1007/s00018-009-0109-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0109-0