
Abstract
Membrane-embedded β-barrel proteins span the membrane via multiple amphipathic β-strands arranged in a cylindrical shape. These proteins are found in the outer membranes of Gram-negative bacteria, mitochondria and chloroplasts. This situation is thought to reflect the evolutionary origin of mitochondria and chloroplasts from Gram-negative bacterial endosymbionts. β-barrel proteins fulfil a variety of functions; among them are pore-forming proteins that allow the flux of metabolites across the membrane by passive diffusion, active transporters of siderophores, enzymes, structural proteins, and proteins that mediate protein translocation across or insertion into membranes. The biogenesis process of these proteins combines evolutionary conservation of the central elements with some noticeable differences in signals and machineries. This review summarizes our current knowledge of the functions and biogenesis of this special family of proteins.
Similar content being viewed by others


Bacterial β-barrel proteins
The Gram-negative bacterial cell envelope
Gram-negative bacteria are enveloped by two membranes, the inner and the outer. The space between these membranes, the periplasm, contains the cell wall, which consists of peptidoglycan. While the inner membrane is a regular phospholipid bilayer, the outer membrane (OM) is an asymmetrical bilayer consisting of phospholipids and lipopolysaccharides (LPS) in the inner and outer leaflets, respectively. Furthermore, the integral membrane proteins of the inner and outer membrane are structurally different. While integral inner membrane proteins typically span the membrane in the form of hydrophobic α-helices, most integral outer membrane proteins (OMPs) are β-barrels consisting of antiparallel amphipathic β-strands [1] (see Fig. 1 for an example of the structure of a membrane-embedded β-barrel protein). A notable exception is Wza, the OM transporter for capsular polysaccharides. Each protomer of this octameric protein contributes an amphipathic α-helix to a barrel of α-helices that is embedded in the membrane [2]. The general absence of hydrophobic α-helices in OMPs is probably related to the fact that they have to be transported across the inner membrane to reach their destination; they would be retained in the inner membrane if they contained hydrophobic α-helices (see below). Apart from integral membrane proteins, the OM also contains lipoproteins, which are attached to the membrane via an N-terminal N-acyl-diacylglycerylcysteine moiety. For the biogenesis of these lipoproteins, which will not be discussed here, the reader is referred to other reviews [3, 4].

Crystal structure of a bacterial β-barrel outer membrane protein. Left: a ribbon representation of the structure of LpxR, a lipid A deacylase of Salmonella typhimurium [133]. The 12-stranded β-barrel consists of all anti-parallel β-strands, which are connected by short turns at the periplasmic side (bottom) and longer loops including some α-helical segments at the extracellular side (top). The ribbon is colored with a gradient from the N terminus in blue to the C terminus in red. Right a top view of the protein (the figure was kindly provided by Lucy Rutten)
Biogenesis of bacterial outer membrane proteins
OMPs are synthesized in the cytoplasm as precursors with an N-terminal signal sequence (Fig. 2). These precursors are bound by the chaperone SecB, which prevents their aggregation in the cytoplasm and thereby keeps them in a translocation-competent state. The signal sequence and the bound chaperone target the precursors to the Sec machinery in the inner membrane, which translocates these proteins across the membrane into the periplasm where the signal sequence is cleaved off (Fig. 2) [5, 6]. The Sec machinery also deals with integral inner membrane proteins. However, when a hydrophobic transmembrane segment of such a protein is present in the Sec channel, the channel will open laterally to allow for the anchoring of such a segment into the membrane [5, 6]; this is most likely the reason for the absence of such segments in integral OMPs.

Biogenesis of bacterial outer membrane proteins. OMPs are synthesized in the cytoplasm as precursors with an N-terminal signal sequence (SP) (1). Next, they are transported across the inner membrane (IM) to the periplasm via the Sec translocon (2). The holding chaperones SecB and Skp prevent premature folding and aggregation of the OMPs in the cytoplasm and periplasm, respectively. The OMPs are then targeted to the Bam complex in the outer membrane (OM) (3), which consists of the integral membrane protein Omp85 (BamA) and four membrane-associated lipoproteins BamB–E (alternative names indicated on figure in parentheses). Omp85 consists of a C-terminal β-barrel embedded in the membrane and an N-terminal part consisting of five polypeptide-transport-associated (POTRA) domains (P1–P5) extending in the periplasm. The periplasmic chaperone SurA assists in the folding of the OMPs at the Bam complex that assembles them into the outer membrane, where they can reside as monomers or oligomers
Several chaperones that guide nascent OMPs in the periplasm have been identified. The most important of those are ( seventeen-kD protein (Skp) and SurA (Fig. 2) [3]. Skp selectively binds unfolded OMPs [7, 8] early on while they are still engaged with the Sec machinery [9]. The phospholipid-binding properties of Skp [8], which support its localization to the external surface of the inner membrane, probably enable this early interaction. Binding of Skp may assist in the release of the nascent OMP from the Sec machinery and results in the formation of a soluble periplasmic intermediate [10]. Hence, Skp functions as a holding chaperone that prevents aggregation of its substrates in the periplasm. The crystal structure of the trimeric Skp revealed a jellyfish-like architecture with the tentacles forming a cavity where the unfolded substrate proteins could bind [11, 12]. Recently, the interaction between Skp and OmpA, a two-domain OMP consisting of a membrane-embedded β-barrel and a periplasmic peptidoglycan-binding domain, was studied in vitro in biochemical and NMR experiments [13]. These experiments confirmed the working model for Skp function. They revealed that trimeric Skp prevents unfolded OmpA from aggregating by forming stable soluble 1:1 complexes. Within the complex, the β-barrel domain of OmpA remains unfolded and is buried deep within the cavity among the tentacles of Skp, while its periplasmic domain is free to fold and extends away from the complex [13].
An additional chaperone, SurA, was first identified as a protein required for survival of E. coli in the stationary phase [14]. In contrast to Skp, which functions as a holding chaperone, SurA functions as a folding chaperone that assists the folding of nascent OMPs into their native conformation [15, 16]. The 46-kDa protein contains two peptidyl-prolyl isomerase (PPIase) domains, which, however, are dispensable for function [17]. The crystal structure of SurA revealed a globular core fragment, consisting of the N- and C-terminal domains and the first (inactive) PPIase domain with the second (active) PPIase domain extending away from the core domain [18]. The core shows an extended crevice in the N-terminal domain where peptides could bind. Peptide binding studies revealed that SurA has a preference for peptides rich in aromatic residues, particularly those containing two consecutive aromatic residues or two aromatic residues separated by one other residue, i.e., sequences typically found in the transmembrane β-strands of OMPs [19, 20]. Such peptides were subsequently found to be recognized by the first PPIase domain of SurA [21], which is dispensable for function. In contrast, an earlier report suggested that peptide binding is mediated by the N-terminal domain of SurA [22]. This discrepancy can be explained if substrate selection and chaperoning activity are distinct activities that reside in different parts of the protein [21].
Skp and SurA are both non-essential proteins in E. coli as mutants in whom the corresponding genes are inactivated are viable. Such mutants have drastically decreased OMP levels [7, 23]. This reduction is due to the induction of a periplasmic stress response including, amongst others, the over-expression of the potent protease DegP, which degrades unassembled OMPs in the periplasm. Furthermore, the synthesis of small regulatory RNAs that inhibit the translation of the mRNAs for OMPs is induced [24, 25]. The observation that skp and surA mutations are synthetically lethal might indicate that these proteins have overlapping functions and that they operate in the periplasm in parallel pathways for OMP assembly [23, 26]. However, because of their different structure, mode of substrate binding, and function as described above, alternative explanations for this synthetic lethality should be considered. We favor the model that Skp and SurA act sequentially within the same pathway (Fig. 2) [3]. The synthetic lethality in this view is explained by an increased requirement for a holding chaperone when the folding of the substrates is compromised by the absence of SurA, and, vice versa, efficient folding is increasingly important when the holding chaperone Skp is absent. Consistent with this view, Skp acts early within the periplasmic pathway while the substrate proteins are still engaged with the Sec translocon in the inner membrane [9]. In contrast, SurA could be cross-linked to the Bam complex in the OM [23], which is required for assembly of OMPs into the OM (see below), suggesting that SurA acts late in the pathway.
Apart from Skp and SurA, DegP has also been suggested to be involved in OMP biogenesis [23, 27]. DegP is a periplasmic protease, which has chaperone qualities as well [28]. In its activated form, DegP forms large, cage-like 12-meric and 24-meric complexes [27, 29], which could harbor a folded OMP in their cavities [27]. However, so far, there is little evidence for a direct role of DegP in OMP folding or insertion. The synthetic lethality of a degP mutation when combined with a surA [26] or skp [10] mutation is probably not due to its capacity as a chaperone to take over the role of SurA and Skp and assist in OMP assembly, but rather reflects its capacity as a protease to prevent the toxic accumulation of unfolded OMP aggregates in the periplasm. Of note, it is not only the protease activity of DegP that is important in this respect. A mutant form of DegP without protease activity but with retained chaperone activity could rescue strains from lethality caused by the expression of assembly-defective mutant forms of OmpF and OmpC. This rescue occurred without restoring the assembly of these mutant proteins into the OM, but rather by sequestering them, thereby removing them from the assembly pathway [30, 31]. This sequestering role could be reflected by the captured OMPs observed in the DegP multimeric complexes [27]. In conclusion, Skp and SurA are the most important periplasmic chaperones in OMP biogenesis and they appear to perform different roles in this process.
The β-barrel assembly machinery
After passage through the periplasm, OMPs are assembled into the OM via a machinery, recently designated the β-barrel assembly machinery or Bam complex (Fig. 2). The first component of this machinery was identified in Neisseria meningitidis, where it was demonstrated that a highly conserved OMP, designated Omp85, is essential for viability and required for the folding and assembly of all OMPs examined [32]. In a mutant strain, in which the expression of the omp85 gene could be regulated, unfolded forms of OMPs accumulated as periplasmic aggregates when the synthesis of Omp85 was switched off [32, 33]. Similarly, a role in OMP assembly was subsequently demonstrated for the Omp85 homologues of E. coli [34–36] and Pseudomonas aeruginosa [37], designated BamA (formerly YaeT) and Opr86, respectively.
The Omp85 homologue of E. coli, BamA, was found to form a complex with four lipoproteins, designated BamB–E (formerly YfgL, NlpB, YfiO, and SmpA, respectively) [36, 38] (Fig. 2). Of these, only BamD is essential for viability [39] and OMP assembly [40], whereas deletion of the other accessory components, which are evolutionarily less well conserved, does not infringe viability and results in relatively mild OMP assembly defects. Interestingly, the BamD homologue of Neisseria gonorrhoeae, designated ComL, is covalently linked to the peptidoglycan layer [41], while bamB mutations were reported to affect peptidoglycan synthesis, possibly by regulating the activity of lytic transglycosylases [42]. Furthermore, many bacteria contain a BamE homologue extended with a peptidoglycan-binding domain, for example the Plp4 protein in Pasteurella haemolytica [43]. These observations suggest that these accessory components may have a role in anchoring the Bam complex to the peptidoglycan and perhaps in modulating the peptidoglycan to facilitate the access of nascent OMPs to this complex. Such a putative peptidoglycan-related role of these accessory components would explain why similar components are absent from the machinery required to assemble β-barrel proteins in mitochondria, which do not contain peptidoglycan. Curiously, BamB was also reported to have protein kinase activity and to be involved in DNA strand-break repair and homologous recombination [44], but all these properties are difficult to reconcile with its OM-associated periplasmic location.
Substrate recognition by Omp85
The Omp85 proteins of several bacteria, including BamA from E. coli, were found to form pores when reconstituted in planar lipid bilayers or liposomes [45–48]. The physiological significance of these pores is not entirely clear, but this property could be used to study the interaction with substrate proteins, because it was anticipated that such interaction would affect the channel properties. Indeed, it was found that denatured E. coli OMPs, but not periplasmic proteins, increased the BamA channel activity, showing that these substrates directly and specifically interact with BamA [47]. The channel-enhancing activity was mimicked by a synthetic peptide corresponding to the last 12 C-terminal amino-acid residues of the OMP PhoE, suggesting that the C terminus contains the signal for substrate recognition. In agreement with this result, earlier studies have recognized a conserved signature sequence at the C terminus of the vast majority of OMPs from various bacterial species [49]. This signature sequence consists of a highly conserved Phe (or Trp) at the ultimate C-terminal position, Tyr or a hydrophobic residue at position 3, and also hydrophobic residues at positions 5, 7, and 9 from the C terminus. It was demonstrated that the deletion of the conserved C-terminal Phe in PhoE dramatically affected the efficiency of assembly into the OM, and the mutant protein was found to accumulate as dense aggregates in the periplasm [49]. The expression of the mutant protein was lethal. Consistent with these in vivo experiments, it was found that a synthetic peptide corresponding to a C-terminal fragment of PhoE but lacking the C-terminal Phe failed to enhance the BamA channel activity in planar lipid bilayers [47].
Of note, a C-terminal Phe (or Trp) is not an absolute requirement for assembly into the OM. A substitution analysis showed that both the hydrophobicity and the aromatic nature of the C-terminal residue were of importance, but a Tyr or a hydrophobic residue at this position was less detrimental for assembly than the presence of a polar residue [49]. Furthermore, a mutant PhoE protein with a deletion of the C-terminal Phe was efficiently assembled into the OM provided the expression level was reduced [50]. Thus, even though the removal of the C-terminal Phe reduces efficient recognition by the assembly machinery, it does not completely destroy the recognition signal, or, alternatively, less efficient signals in other locations within the PhoE sequence also exist. Consequently, at high expression levels, the mutant protein will aggregate within the periplasm because of the poor recognition by the assembly machinery. The reduced kinetics of aggregation at lower expression levels, however, increases the time span for the mutant protein to functionally interact with the Bam complex and result in assembly of the mutant protein.
Although the C-terminal signature sequence described above is present in the vast majority of OMPs from various bacterial species, some species-specific adaptations appear to have occurred during evolution. In the vast majority of OMPs of Neisseria spp., a positively charged residue is present at the penultimate position from the C terminus, whereas this is seldom the case in E. coli. High-level expression of these OMPs is lethal in E. coli, and these OMPs fail to activate the BamA channels in planar lipid bilayers. Substitution of this positively charged residue by a glutamine, which is often found at this position in E. coli OMPs, markedly improved the assembly of the meningococcal porin PorA into the E. coli OM [47].
Structure of Omp85
Omp85 was suggested to consist of two domains [32], a C-terminal membrane-embedded β-barrel and an N-terminal periplasmic part (Fig. 2). The latter part consists of five repeated sub-domains, known as polypeptide-transport-associated (POTRA) domains [51]. The function of the different POTRA domains is not entirely clear, although, considering their periplasmic location, a role in the binding of substrates and/or the accessory components of the Bam complex is likely. In N. meningitidis Omp85, the first four POTRA domains could be deleted simultaneously resulting in only relatively mild assembly defects particularly of large OMPs [52]. Only the fifth POTRA domain together with the β-barrel domain was found essential in this study. Hence, the functional core domain of the bacterial Omp85 is similar to that of its mitochondrial homologue Sam50/Tob55, which consists of only a single POTRA domain attached to a β-barrel and will be discussed later. In contrast, deletion of each individual POTRA domain, except for the second one, of E. coli BamA severely infringed function and viability of the cells [53]. Apparently, there are considerable differences in Omp85 functioning between different bacteria. Deletion of the fifth POTRA domain in E. coli BamA resulted in the loss of binding of BamB–E [53], which suggests a specific role for this POTRA domain in the binding to the accessory lipoproteins and is consistent with the important role of this domain in N. meningitidis Omp85.
Recently, the structure of the POTRA domains of E. coli BamA was investigated by X-ray crystallography [53, 54], NMR spectroscopy, and small-angle X-ray scattering [55]. Each POTRA domain consists of a three-stranded β-sheet overlaid with a pair of α-helices, and the linkers between the domains appear to allow some flexibility in their relative orientation. Interestingly, OmlA, the BamE homologue of Xanthomonas axonopodis pv. citri, has a similar structure, although the relative order of the secondary structure elements is different [56]. The crystallization-induced dimerization of a polypeptide containing the first four POTRA domains of BamA and a small part of the fifth one suggested that the POTRA domains might bind substrates and/or accessory proteins by a process called β-augmentation [53]. NMR experiments revealed that various peptides of the model OMP PhoE could bind, although weakly, to either side of the β-sheets of the POTRA domains, and suggested that the POTRA domains could guide nascent OMPs to the core of the Bam complex by processive sliding [55]. Unfortunately, binding of a C-terminal peptide of PhoE or another OMP could not be measured in these experiments because of solubility problems.
The structure of the β-barrel of an Omp85 protein has so far not been solved. However, the Omp85 super-family also includes bacterial OMPs involved in the two-partner secretion pathway. These proteins, generically called TpsB, are required for the translocation of their partner, TpsA, across the OM into the medium. Recently, the structure of a TpsB protein, i.e., FhaC of Bordetella pertussis, was solved [57]. The structure showed that the protein consists of a periplasmic moiety containing two POTRA domains and a 16-stranded C-terminal β-barrel embedded in the OM. The pore in the barrel is closed by an N-terminal α-helix and by a loop that reaches from the cell-surface-exposed side through the barrel to the periplasmic side of the membrane. It is plausible that the removal of the helix and the loop from the barrel upon substrate binding would create a pore allowing for the transport of the partner TpsA to the cell surface. Unfortunately, this structure provides little insight into how the β-barrel of Omp85 might work, as it is not conceivable that nascent OMPs would insert into the barrel of Omp85 [58]. Rather, membrane insertion should take place at the subunit interface if Omp85 forms oligomers, for which some evidence has been reported [47], or at the lipid–protein interface. Further discussion on putative mechanisms for membrane insertion is included in the section on the biogenesis of mitochondrial β-barrel proteins.
Mitochondrial β-barrel proteins
Functions and structural features of eukaryotic β-barrel proteins
Compared to the diversity of β-barrel membrane proteins in prokaryotes, the number of mitochondrial OMPs belonging to this structural type is rather limited. Although some prediction programs propose that the yeast mitochondrial OM might contain more than 100 β-barrel proteins [59], later proteomics and bioinformatic studies revealed that this number is an overestimation. In the extensively studied unicellular fungus Saccharomyces cerevisiae, six members have been identified: Tom40, Tob55/Sam50, two isoforms of porin/VDAC, Mdm10, and Mmm2, of which only the first two are essential for viability [60, 61]. It should be noted that, to date, only the structure of mammalian VDAC1 has been solved and the remaining members have been assigned to the family on the basis of both secondary structure content and in silico predictions.
Mitochondrial porin (also named voltage-dependent anion-selective channel, VDAC) is the founding member of the eukaryotic family [62]. VDAC is the most abundant protein in the mitochondrial OM and, soon after its discovery more than three decades ago, its role as a diffusion pore for small metabolites became apparent [63]. Conductivity and selectivity of the pore are achieved by a gating mechanism, which can be influenced both in vitro and in vivo by the membrane potential and by a variety of binding partners (reviewed in [64]). In yeast, two isoforms, VDAC1 and VDAC2, encoded by the genes POR1 and POR2, respectively, are known. Deletion of POR1 resulted in significant growth phenotypes whereas VDAC2 appears to be dispensable. However, the observation that over-expression of VDAC2 could partially complement the Δpor1 phenotype [65] suggests similarities in their function. In mammals, the family of VDAC proteins comprises three known orthologues, all of them mapping to individual gene loci [66, 67]. The third isoform, VDAC3, which is assumed to be the most primordial member [67], was found to yield an additional variant via tissue-specific alternative splicing [68]. In cultured cells, none of the three isoforms appeared to be essential when deleted individually, but mitochondrial respiration was reduced [69].
VDAC is located on the interface between mitochondria and the cytosol and thus has the potential to act as a “switch” for cellular processes involving mitochondria. This position and its central role in mitochondrial permeability define VDAC as a future pharmacological target for anticancer agents [70]. Indeed, an increasing body of evidence has been collected in support of a role of VDAC1 in apoptosis-mediated cell death in higher eukaryotes. It was suggested that the protein participates in the mitochondrial permeability transition (MPT) and in the exit channel for the release of cytochrome c from the mitochondrial intermembrane space (IMS) into the cytosol. This release is enhanced by pro-apoptotic Bax/Bak proteins and inhibited by anti-apoptotic Bcl-xL [71–73]. However, the physiological importance of VDAC proteins to apoptosis was challenged by the finding that fibroblasts lacking all three VDAC isoforms exhibited MPT and cell death indistinguishable from wild-type cells [74]. In addition to its proposed interactions with pro- and anti-apoptotic proteins, various cytosolic proteins (like hexokinase and tubulin) and characteristic mitochondrial lipids (like phospatidylethanolamine and cardiolipin) were suggested to regulate VDAC gating [75]. Taken together, although it is evident that VDAC is playing a key role in the communication between mitochondria and the rest of the cell, future studies are required to shed more light on the regulation of its activity and the integration of such activity in various signaling processes.
Tom40 was the second member of the mitochondrial β-barrel OMP family to be identified. It is essential in both S. cerevisiae and Neurospora crassa and was found to be the central component of the translocase of the mitochondrial OM (TOM complex) [76, 77]. Tom40 is required for the import of the vast majority of precursor proteins into mitochondria as it forms the general import pore of the complex [78, 79]. The second essential protein in the mitochondrial OM is Tob55 (also termed Sam50 [80] or Omp85 [81]). The protein is the central member of a complex termed TOB (for topogenesis of β-barrel proteins [82]) or SAM (for sorting and assembly machinery [80]). Notably, Tob55 is conserved in all eukaryotes and is the only mitochondrial β-barrel protein that shares significant sequence homology with β-barrel proteins from chloroplasts (Toc75-V) and bacteria (Omp85/BamA) [81, 82]. Tob55 promotes the membrane integration of precursors of β-barrel proteins and its function will be discussed in detail below.
The two additional mitochondrial β-barrel proteins in fungi, Mdm10, and Mmm2 (Mdm34) are required for mitochondrial morphology and dynamics [83, 84]. Homologues of these two proteins have so far not been reported in mammals. The precise role of Mmm2 is not clear. Mdm10 was suggested to be part of a complex in the OM that links mitochondrial membranes and DNA to the cytoskeleton-based segregation machinery [85]; on the other hand, it was also proposed that Mdm10 is part of the TOB complex and has a role in the biogenesis of β-barrel proteins [86]. This second putative role is discussed in a later section.
More than 25 atomic structures of bacterial β-barrel proteins led to the consensus that such proteins are composed of an even number of β-strands arranged in an antiparallel manner. The recently solved first atomic structure of a mammalian β-barrel protein, VDAC1, changed this view. Three independent studies using NMR spectroscopy, X-ray crystallography, or a combined approach revealed that the barrel structure of VDAC1 comprises 19 β-strands, which are interconnected by relatively short turns between 2 and 10 amino-acid residues in length, in an antiparallel orientation [87–89]. Hence, unlike all known prokaryotic β-barrel proteins, the structure is formed by an odd number of β-strands. Surprisingly, a parallel β-sheet formed between strands 1 and 19 was detected. It has been reported that a mutant form of the E. coli porin PhoE lacking the first transmembrane β-strand could be functionally incorporated into the bacterial OM [90], showing that the bacterial Bam machinery can also deal with barrels with an odd number of strands. A short N-terminal helix was resolved in the centre of the VDAC1 channel. This helix was proposed to have a function in the transition between open and closed states [89]. Based on computational analysis, it was suggested that the topology of VDAC1 could also apply to other VDAC isoforms and to Tom40 [87]. Being the first structure of a eukaryotic β-barrel membrane protein, additional structures are required to understand whether the unique structural features of VDAC1 are shared by other eukaryotic proteins.
Biogenesis of β-barrel proteins in mitochondria
A comparison between the biogenesis pathways of β-barrel proteins in mitochondria and bacteria has several implications regarding the evolutionary conservation of the process. Besides differences in the protein composition between the Bam and TOB complexes, the compartment in which the precursors of β-barrel proteins are synthesized has in the case of mitochondria shifted to the exterior of the organelle, most likely due to gene transfer from the endosymbiont’s genome to the nucleus. Intriguingly, in spite of the direct accessibility of the mitochondrial OM to cytosolic β-barrel precursors, a situation not found in bacteria, there is no direct membrane insertion from the cytosolic face of the OM, but rather these precursors are transported into the IMS before assembly. Thus, in both systems, the precursors are inserted from the internal side of the outer membrane. This conservation is also reflected in the orientation of the central component of the Bam/TOB machinery where in both cases the POTRA domain(s) points toward the IMS/periplasm.
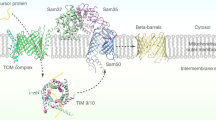
Newly synthesized mitochondrial β-barrel proteins are initially recognized by the receptor components of the TOM complex, Tom20 and Tom70. They are then translocated through the import pore of the TOM complex and relayed to the TOB/SAM complex, which mediates their insertion into the OM. On their way from the TOM to the TOB complex, the precursor proteins are exposed to the IMS where they interact with small Tim components (Fig. 3). A major part of our knowledge about the biogenesis of β-barrel proteins results from studies on Tom40, the key component of the TOM complex. A major advantage for studying the protein’s assembly lies in the fact that several distinct assembly intermediates can be followed over time when radiolabeled Tom40 precursors are imported into isolated mitochondria and subsequently analyzed by Blue Native PAGE (reviewed in [60, 61]). In the following sections, steps in the biogenesis of β-barrel proteins and the components involved will be discussed.

Working model for the biogenesis of mitochondrial β-barrel proteins. Precursors are synthesized in the cytosol and are probably guided to the mitochondrial surface by chaperones (1). At the mitochondrial surface, precursors are engaged by the primary import receptors of the TOM complex, predominantly via an interaction with Tom20. Subsequently, they are translocated through the TOM pore across the mitochondrial outer membrane (OM) into the intermembrane space (IMS) (2), where they interact with the small Tim protein complexes, which exert a chaperone-like function (3). Through a second step of recognition, the precursor proteins are sorted to the TOB complex, which then promotes their integration into the lipid core of the outer membrane (4)
Targeting information of mitochondrial β-barrel proteins
Other than matrix-destined proteins, β-barrel precursors are devoid of canonical N-terminal targeting sequences [91]. In-depth analysis of the insertion pathway has led to the understanding that the correct sorting to its destination involves a sequence of at least two recognition steps. First, after its synthesis in the cytosol, the precursor protein is recognized at the TOM complex similar to virtually all other mitochondrial proteins [92–95]. This recognition is followed by translocation across the import pore of the TOM complex and thus assures import into mitochondria and therefore targeting to the correct organelle. Second, precursor-sorting pathways diverge, and an interaction with the small Tim chaperones followed by recognition by the TOB machinery assures a distinct suborganellar sorting of β-barrel precursors to the OM. The signals required for recognition in each of these two steps are believed to differ in their nature.
Early studies based on in vitro import of mutated radiolabelled β-barrel precursors into isolated mitochondria failed to conclusively locate specific sequences responsible for mitochondrial targeting. For example, C- or N-terminal truncations of N. crassa Tom40 by 20 or 60 amino-acid residues, respectively, failed to abolish import [94]. In the case of the precursor of N. crassa porin, only the N terminus was dispensable for targeting whereas the C-terminal 15 amino-acid residues were required [96]. Likewise, various point mutations as well as terminal or internal deletions in yeast porin could abolish precursor import [96, 97]. Furthermore, in contrast to matrix-destined proteins, unfolding of Tom40 precursors resulted in decreased import and assembly efficiency [94]. These findings indicate that the signal for targeting of β-barrel precursors to mitochondria is probably not confined to a single linear sequence but rather involves structural elements.
Several reports have led to the assumption that a precursor’s ability to form a β-barrel structure can be sufficient to ensure its targeting to the mitochondrial compartment. A first hint was provided by studies on the role of the bacterial OMP PorB from pathogenic Neisseria spp. during infection. After invasion, the protein is released into the host cell’s cytosol from where it is translocated into mitochondria and induces cell death via the intrinsic apoptotic pathway [98, 99]. Very recently, we could demonstrate that PorB is not an exceptional case and other bacterial OMPs share the ability to be targeted to mitochondria when expressed in yeast cells. In contrast to PorB in mammalian cells, however, production of these proteins caused neither mitochondrial dysfunction nor other signs of cellular damage. Like endogenous β-barrel proteins, the bacterial members were inserted in a process that was found to depend on both TOM- and TOB-complex components and even resulted in the formation of native-like oligomeric structures in the mitochondrial OM [95]. Taken together, we speculate that, rather than eukaryote-specific linear signals, a folding intermediate of a precursor with high β-sheet content can serve as a signal of β-barrel proteins to assure their targeting from the cytosol to their destined compartment. The targeting issue becomes even more complicated in plant cells that contain two organelles with β-barrel membrane proteins, mitochondria and chloroplasts. The question how these cells assure that β-barrel proteins reach only one of these organelles is still open.
While recognition by and translocation through the TOM complex is shared among the majority of mitochondrial protein precursors, the sorting pathways diverge after the translocation step, depending on their destination within the organelle. In the case of β-barrel proteins, this second recognition step involves an interaction between its C terminus and the TOB machinery. Very recently, a signature motif termed β-signal, located at the C-terminal β-strand of the precursor, was identified [100]. This β-signal is different, although possibly derived from the C-terminal signature sequence found in bacterial OMPs and described above. Through site-directed mutagenesis, four essential residues were identified of which three, two hydrophobic and one hydrophilic, were found to be essential for binding to the TOB complex. In contrast, mutation of a conserved glycine resulted in the accumulation of TOB complex-associated assembly intermediates, indicating a role in release from the complex. Sequence analysis revealed that the identified β-signal is conserved in mitochondrial β-barrel proteins from organisms from all eukaryotic kingdoms: fungal species, plants, and animals including humans [100].
Components of the TOB complex
The known members of the TOB core complex in the fungal model organism S. cerevisiae are Tob55/Sam50/Omp85, Tob38/Sam35, and Mas37/Tom37/Sam37 (Fig. 3). Mas37 was initially identified in a screen for mutants defective in the control of phospholipid biosynthesis. However, due to genetic interaction with Tom20 and Tom70, it was proposed to function as an import receptor of the TOM complex [101]. Later, its membership in a distinct complex and its role in β-barrel precursor insertion became apparent [102]. Unlike the other two TOB complex components, Mas37 is not required for viability unless cells are grown at elevated temperatures [101].
The second member of the complex to be characterized was Tob55, which was identified by several independent approaches. First, both a proteomic screen of vesicles derived from the mitochondrial OM of N. crassa [82] and genome-wide in silico homology searches [81] revealed the presence of a protein with significant sequence similarity to the previously characterized Omp85, which facilitates OM β-barrel protein insertion in N. meningitidis [32]. Second, Tob55 co-purified with affinity-tagged Mas37 [80]. Like its bacterial ancestors from the Omp85 family, Tob55 comprises a membrane-embedded C-terminal β-barrel domain. Its N terminus forms a soluble extension towards the IMS, i.e., a POTRA domain [51]. While all bacterial Omp85 proteins contain five of these domains, only a single one exists in the eukaryotic orthologues. In conditional mutants [80, 81] and in a strain in which Tob55 was depleted via an inducible promoter [82], a selective reduction in the steady-state levels of known β-barrel proteins in the mitochondrial OM was observed. Deletion of Tob55 in yeast is lethal, underscoring its crucial role in the function of the TOB complex.
The second essential component of the TOB complex, Tob38, was identified by co-purification with Mas37 [103] or Tob55 [104]. Furthermore, it was observed that β-barrel assembly was impaired when the protein was depleted or mutated [103–105]. The fact that the protein is both extractable under alkaline conditions and degradable by treatment of intact mitochondria with protease led to the proposal that it is not embedded within the membrane but rather associated with the complex on the cytosolic face of the mitochondrial OM. A recent study indicated that the vast majority of the polypeptide chain is located within the membrane-embedded part of the TOB complex and only the termini are exposed to the cytosol [100].
Of note, while Tob55 shows clear homology with the bacterial Omp85 proteins, there is no sequence similarity of Mas37 or Tob38 with any of the accessory lipoproteins from the bacterial Bam complex. The degree to which the mitochondrial proteins are conserved between different eukaryotic kingdoms varies strongly between the individual members of the TOB complex. Clearly, orthologues of Tob55 with a high degree of sequence similarity exist in all eukaryotic genomes sequenced to date. Although clear homologues of Mas37 are only present in fungal species, it was suggested that mammalian Metaxin 1 may represent a distant orthologue as both proteins show sequence similarity in their N-terminal regions [106]. However, unlike Mas37, which is peripherally associated on the cytosolic face of the mitochondrial OM, Metaxin 1 is probably anchored to the OM via a transmembrane segment at its C-terminal domain. The importance of this protein is reflected by the early embryonic lethal phenotype of mice lacking Metaxin 1 [106]. Later, a related protein named Metaxin 2, which shares 29% identity with Metaxin 1, was identified [107]. Despite very weak homology, Metaxin 2 was suggested to be related to Tob38 [103, 104]. This notion is supported by the observation that both Metaxins interact with each other [107] and play a role in the assembly of Tom40 and VDAC [108].
It is still a matter of debate whether an additional β-barrel protein, Mdm10, is a further component of the TOB complex. Deletion of Mdm10 results in abnormal mitochondrial morphology [83], and the protein was further shown to exist in a complex with Mmm1 and Mdm12, which facilitates attachment of actin fibres to the organelle [85]. The picture became complicated when it was found that Mdm10 also functions in the assembly of Tom40 but is not involved in the biogenesis of other β-barrel proteins [86]. A later study suggested that Mmm1/Mdm12 function in the major β-barrel pathway, which involves all known β-barrel precursors, in a stage downstream of the TOB complex [109]. Currently, it is unclear whether the primary function of these proteins lies in mediating the organelle’s dynamics, in protein sorting, or whether they have dual function.
Assembly of the TOB complex
The TOB complex, comprising all three known constituents, Tob55, Tob38, and Mas37, has a molecular weight of 200–250 kDa as determined by size-exclusion chromatography and Blue Native PAGE. In contrast, a sub-complex lacking Mas37 has a molecular weight of approximately 160 kDa [80, 104, 105]. Since β-barrel assembly is possible in mas37Δ strains, it is likely that the Tob55-Tob38 sub-complex represents a minimal functional unit.
The biogenesis pathways by which the TOB complex constituents are incorporated into functional machinery vary between the individual members. Tob55, being a β-barrel protein on its own, is inserted in the same way as the other members of this structural family. Its C-terminal β-barrel domain contains the necessary targeting information and is both required and sufficient for assembly into the complex while the N-terminal POTRA domain is required neither for targeting to the organelle nor for intra-mitochondrial sorting [110]. After their synthesis in the cytosol, Tob55 precursors are imported into the IMS of mitochondria via the TOM translocation pore and further integrated into the mitochondrial OM via the TOB complex [111]. Newly imported Tob55 molecules probably assemble into pre-existing TOB complexes.
The targeting of Mas37 precursors to mitochondria and their subsequent incorporation into the TOB complex represent a unique pathway for OMPs, as it is independent of the TOM complex. Tob55 and Tob38 are required for Mas37 assembly whereas the process is even more efficient in mitochondria from a Mas37-deficient strain [111]. Collectively, it appears that the cytosolic precursor protein binds directly to the TOB core complex. So far, the biogenesis pathway of Tob38 has not been studied. However, considering the fact that Tob38 and Mas37 share a comparable topology, one can speculate that a similar pathway applies to both proteins.
Function of the TOB complex
Like the vast majority of mitochondrial proteins, β-barrel precursors are encoded in the nucleus and synthesized in the cytosol on free ribosomes. The delivery to the surface of mitochondria most likely involves cytosolic chaperones in order to keep the precursor proteins in an import-competent partially folded conformation [94]. The precursor proteins are engaged on the organelle surface by import receptors of the TOM complex. This process is mainly dependent on Tom20 while Tom70 only plays a minor role [92–95, 112]. After the initial recognition, β-barrel precursors are translocated through the translocation pore of the TOM complex into the IMS. This conclusion is based on several observations. First, assembly relies on functional Tom40, which is the key component of the TOM pore [92–94]. Second, import can be outcompeted by addition of an excess of matrix-destined precursor proteins, which use the import pore [82, 104, 111]. Finally, assembly intermediates downstream of the TOM complex are protected from externally added proteases [82, 93, 94].
The translocated precursors are then transferred from the TOM to the TOB complex in a process that exposes them to the IMS. At this stage, the small TIM complexes, Tim8/Tim13 and Tim9/10, which reside in this compartment, stabilize the β-barrel precursors [111, 113, 114]. Such an involvement of the small Tims can explain early observations where rupturing of the OM was found to impair porin biogenesis in vitro [115]. Like Skp in the bacterial systems, the small TIM complexes are believed to function as chaperones that prevent precursor aggregation or misfolding in the IMS, and they were also shown to be involved in the biogenesis of hydrophobic inner membrane proteins [116, 117].
Several lines of evidence suggest that translocation through the TOM pore and membrane insertion by the TOB complex do not occur separately but are rather coupled processes. When TOB complex function is reduced by deletion of Mas37 or depletion of Tob55, no accumulation of precursors in the IMS was observed, but instead, import was reduced to a similar extent as assembly [82, 102]. Furthermore, as discussed above, rupturing of the outer membrane of isolated mitochondria caused a reduction in the assembly of β-barrel precursors. Under these conditions, where externally added precursor proteins have direct access to the TOB complex, blocking the TOM pore resulted in a further inhibition of the already hampered membrane integration efficiency of these precursor proteins [110]. Collectively, these findings indicate that the passage of β-barrel precursors through the TOM pore is probably required in order to yield an insertion-competent state, which can subsequently be processed by the TOB complex. A coupling between the two complexes is further supported by a recent study where bacterial β-barrel precursors were overproduced in yeast cells. Under these conditions, the majority of the bacterial protein was imported but aggregated in the IMS, most likely due to overcharging the insertion capacity of the TOB machinery. Strikingly, under these conditions, import strongly depended on the TOB complex constituents Tob55 and Mas37 [95]. These findings suggest that a functional TOB complex is required for the translocation of β-barrel precursors through the TOM pore. Of note, despite these functional interactions, a direct physical interaction between the TOB and the TOM complex has not yet been observed.
Which parts of the TOB complex recognize and interact with β-barrel precursors is still debateable. Similar to the bacterial Omp85/BamA, Tob55 in all eukaryotes has a POTRA domain in its IMS-located N-terminal domain. In the bacterial Omp85/BamA proteins, at least one of the five POTRA domains is essential for viability, and it was speculated that this domain might interact directly with the bacterial C-terminal signature motif in substrate proteins [52, 53]. Accordingly, yeast cells harboring a Tob55 variant lacking the POTRA domain exhibited slower growth, and a recombinant POTRA domain could bind β-barrel precursor proteins [110]. The importance of the POTRA domain was challenged in a later study where a deletion of the POTRA domain together with some additional residues did not cause any growth phenotype [100]. Furthermore, Kutik et al. identified Tob38 as the component that is responsible for binding the C-terminal sorting signal in β-barrel precursors, termed β-signal [100], which contrasts with the bacterial system where the C-terminal signature sequence of nascent OMPs has been shown to interact directly with the Tob55 homologue Omp85/BamA [47]. It was therefore suggested that, upon precursor binding, Tob38 induces a widening of the TOB complex cavity, allowing a precursor to enter, fold into its β-barrel structure and subsequently be released laterally into the membrane. For this release, a conserved glycine residue within the β-signal appears to be particularly important and its mutation resulted in the accumulation of assembly intermediates in which precursors were bound to the TOB complex [100]. Considering all available data, we propose that the POTRA domain might have an accessory function in intraorganellar sorting of β-barrel precursors, but this function can become redundant due to efficient precursor recognition via Tob38.
The subsequent process in which β-barrel precursors are inserted into the OM is currently unresolved, either for bacterial or for mitochondrial β-barrel proteins. Presumably, in both cases, the integration of all β-strands of the barrel structure into the lipid phase occurs in a concerted manner [118]. It has often been observed that unfolded β-barrel proteins spontaneously insert in vitro into artificial lipid bilayers devoid of any pre-existing proteins [118]. However, the efficiency of this process is low as compared to that in a cellular environment and can be increased by the presence of proteins in the artificial membrane. For example, the reconstitution of either Tom20 or porin into phospholipid membranes enhanced the insertion of other porin molecules into these membranes [112, 119].
Considering the current knowledge, it is tempting to suggest that both the TOB and the Bam machineries catalyze the insertion of β-barrel proteins by functioning as a scaffold element [58, 60, 61]. This proposal is in line with electron micrographs of native isolated TOB complexes where ring-shaped structures measuring approximately 15 nm were predominantly observed. In their centre, these assemblies harbored a central cavity with a diameter of 4–5 nm, which would be sufficient in size to accommodate folded barrel structures of up to 22 β-strands. It was proposed that such a central cavity could function as an Anfinsen-type folding cage similar to chaperonins, allowing the β-barrel structure to form in a protected environment [82]. This relatively large central pore is probably formed among several Tob55 molecules rather than by an individual β-barrel of Tob55. From such a central channel, the precursor protein can be laterally released into the lipid phase of the membrane [100]. As was discussed above regarding the bacterial BamA-mediated membrane insertion, a release of a β-barrel precursor that involves a lateral opening of a single Tob55 β-barrel structure is rather unlikely, since it is thermodynamically unfavorable due to the high number of hydrogen bonds formed between the neighboring membrane-spanning β-strands [120, 121].
Whereas some progress has been made regarding the functions of Tob55 and Tob38, the role of Mas37 in the biogenesis of β-barrel proteins is still ill-defined. It appears that Mas37 has at least two functions. First, it stabilizes in an unknown manner the TOB complex, and in its absence the steady-state levels of the remaining Tob55-Tob38 sub-complex are reduced [104]. Second, Mas37 promotes the dissociation of substrate proteins from the TOB complex. The latter function is supported by the observation that deletion of the protein has a lower impact on the biogenesis of Tob55 as compared to that on other β-barrel proteins [111]. This difference may result from the necessity of most β-barrel proteins to leave the TOB complex before their final integration into the outer membrane, whereas precursor molecules of Tob55 have already reached their destination upon association with pre-existing partially assembled TOB complexes. Furthermore, Chan and Lithgow reported that over-expression of MAS37 can rescue a temperature-sensitive allele of TOB38 without affecting the amount of β-barrel precursors bound to the remaining TOB core complex. Therefore, they suggested that Mas37 acts in β-barrel assembly downstream of Tob38 [122].
β-barrel membrane proteins of chloroplasts
In addition to the OMs of Gram-negative bacteria and mitochondria, β-barrel proteins are also found in the OM of plastids. An in silico approach predicted the presence of many β-barrel proteins in the chloroplast OM [123]. Several of them (for example, OEP16, OEP21, OEP24, and OEP37) were suggested to function as solute channels and have been functionally characterized in conductivity measurements. They represent high-conductance solute channels and their distinct substrate specificities may indicate separate roles in different metabolic processes [124, 125]. In addition, the β-barrel protein Toc75 forms the protein-conducting channel of the translocase of the OM of chloroplasts (TOC complex) [126]. The Toc75 proteins of chloroplasts and cyanobacteria have been annotated as a discrete branch of the Omp85 and Tob55 family [81, 127]. In Arabidopsis thaliana, five different isoforms (I–V) of Toc75 have been identified [128]. Toc75-III is the pore-forming subunit of the TOC complex and, thus, is functionally equivalent to the mitochondrial Tom40 protein [126]. The Toc75-V isoform (OEP80) is a member of a sub-branch, the members of which are closely related to both the SynToc75 proteins of cyanobacteria and the proteobacterial Omp85 and eukaryotic Tob55 proteins [81]. Recently, it was shown that Toc75-V is essential for viability in Arabidopsis [129].
Currently, relatively little is known about the biogenesis pathways of β-barrel proteins in chloroplast. Specific signals for targeting of most β-barrel proteins to the chloroplast are still unknown. An interesting exception is provided by the unique import pathway of the precursor of Toc75-III. Toc75-III is synthesized with an N-terminal extension, which functions as a bipartite transit peptide and is processed during maturation [130]. The first portion of the targeting signal directs the precursor protein to the chloroplasts stroma where it is cleaved by a stromal processing peptidase [131]. The second part probably functions as a stop-transfer segment and was found to be processed by a type I signal peptidase [132]. Thus, currently, Toc75-III is the only known protein in the OM of chloroplasts or mitochondria with a cleavable targeting sequence. Its overall import pathway seems to support the idea that sorting of β-barrel membrane proteins of chloroplasts occurs in a manner similar to that of mitochondria. The Toc75-III precursor is first completely translocated across the outer envelope by the TOC complex and, thus, is likely inserted from the inner face into the lipid phase of the OM. Whether Toc75-V (OEP80) mediates the membrane integration of chloroplast OM β-barrel proteins is an, as yet, unanswered question. In any case, it is not a component of the TOC complex [128]. Surprisingly, the N-terminal region of AtToc75-V (OEP80) is not essential for the targeting, biogenesis, or functionality of the protein suggesting that this protein may follow a different targeting pathway [129].
A central question to be addressed in further studies is how plant cells avoid mis-targeting of β-barrel precursors between mitochondria and chloroplasts. As discussed in the previous sections, small chaperones in the periplasm of bacteria and the IMS of mitochondria were found to be involved in the biogenesis of β-barrel proteins. A challenge for future studies is to find out whether chaperones located between the two chloroplast-envelope membranes are similarly involved in the biogenesis of chloroplast β-barrel proteins. Taken together, although experimental evidence is still lacking, it is tempting to speculate that the principles of β-barrel biogenesis have been conserved from a cyanobacterium to chloroplasts during evolution.
References
Koebnik R, Locher KP, Van Gelder P (2000) Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol 37:239–253
Dong C, Beis K, Nesper J, Brunkan-Lamontagne AL, Clarke BR, Whitfield C, Naismith JH (2006) Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane protein. Nature 444:226–229
Bos MP, Robert V, Tommassen J (2007) Biogenesis of the gram-negative bacterial outer membrane. Annu Rev Microbiol 61:191–214
Tokuda H, Matsuyama S (2004) Sorting of lipoproteins to the outer membrane in E. coli. Biochim Biophys Acta 1693:5–13
de Keyzer J, van der Does C, Driessen AJM (2003) The bacterial translocase: a dynamic protein channel complex. Cell Mol Life Sci 60:2034–2052
Papanikou E, Karamanou S, Economou A (2007) Bacterial protein secretion through the translocase nanomachine. Nat Rev Microbiol 5:839–851
Chen R, Henning U (1996) A periplasmic protein (Skp) of Escherichia coli selectively binds a class of outer membrane proteins. Mol Microbiol 19:1287–1294
De Cock H, Schäfer U, Potgeter M, Demel R, Müller M, Tommassen J (1999) Affinity of the periplasmic chaperone Skp of Escherichia coli for phospholipids, lipopolysaccharides and non-native outer membrane proteins. Role of Skp in the biogenesis of outer membrane protein. Eur J Biochem 259:96–103
Harms N, Koningstein G, Dontje W, Müller M, Oudega B, Luirink J, de Cock H (2001) The early interaction of the outer membrane protein phoE with the periplasmic chaperone Skp occurs at the cytoplasmic membrane. J Biol Chem 276:18804–18811
Schäfer U, Beck K, Müller M (1999) Skp, a molecular chaperone of gram-negative bacteria, is required for the formation of soluble periplasmic intermediates of outer membrane proteins. J Biol Chem 274:24567–24574
Korndörfer IP, Dommel MK, Skerra A (2004) Structure of the periplasmic chaperone Skp suggests functional similarity with cytosolic chaperones despite differing architecture. Nat Struct Mol Biol 11:1015–1020
Walton TA, Sousa MC (2004) Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol Cell 15:367–374
Walton TA, Sandoval CM, Fowler CA, Pardi A, Sousa MC (2009) The cavity-chaperone Skp protects its substrate from aggregation but allows independent folding of substrate domains. Proc Natl Acad Sci USA 106:1772–1777
Tormo A, Almirón M, Kolter R (1990) surA, an Escherichia coli gene essential for survival in stationary phase. J Bacteriol 172:4339–4347
Lazar SW, Kolter R (1996) SurA assists the folding of Escherichia coli outer membrane proteins. J Bacteriol 178:1770–1773
Rouvière PE, Gross CA (1996) SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev 10:3170–3182
Behrens S, Maier R, de Cock H, Schmid FX, Gross CA (2001) The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J 20:285–294
Bitto E, McKay DB (2002) Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10:1489–1498
Bitto E, McKay DB (2003) The periplasmic molecular chaperone protein SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J Biol Chem 278:49316–49322
Hennecke G, Nolte J, Volkmer-Engert R, Schneider-Mergener J, Behrens S (2005) The periplasmic chaperone SurA exploits two features characteristic of integral outer membrane proteins for selective substrate recognition. J Biol Chem 280:23540–23548
Xu X, Wang S, Hu YX, McKay DB (2007) The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues. J Mol Biol 373:367–381
Webb HM, Ruddock LW, Marchant RJ, Jonas K, Klappa P (2001) Interaction of the periplasmic peptidylprolyl cis–trans isomerase SurA with model peptides. The N-terminal region of SurA is essential and sufficient for peptide binding. J Biol Chem 276:45622–45627
Sklar JG, Wu T, Kahne D, Silhavy TJ (2007) Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev 21:2473–2484
Johansen J, Rasmussen AA, Overgaard M, Valentin-Hansen P (2006) Conserved small non-coding RNAs that belong to the σE regulon: role in down-regulation of outer membrane proteins. J Mol Biol 364:1–8
Ruiz N, Silhavy TJ (2005) Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr Opin Microbiol 8:122–126
Rizzitello AE, Harper JR, Silhavy TJ (2001) Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J Bacteriol 183:6794–6800
Krojer T, Sawa J, Schäfer E, Saibil HR, Ehrmann M, Clausen T (2008) Structural basis for the regulated protease and chaperone function of DegP. Nature 453:885–890
Spiess C, Beil A, Ehrmann M (1999) A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell 97:339–347
Jiang J, Zhang X, Chen Y, Wu Y, Zhou ZH, Chang Z, Sui SF (2008) Activation of DegP chaperone-protease via formation of large cage-like oligomers upon binding to substrate proteins. Proc Natl Acad Sci USA 105:11939–11944
CastilloKeller M, Misra R (2003) Protease-deficient DegP suppresses lethal effects of a mutant OmpC protein by its capture. J Bacteriol 185:148–154
Misra R, CastilloKeller M, Deng M (2000) Overexpression of protease-deficient DegPS210A rescues the lethal phenotype of Escherichia coli OmpF assembly mutants in a degP background. J Bacteriol 182:4882–4888
Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J (2003) Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299:262–265
Voulhoux R, Tommassen J (2004) Omp85, an evolutionarily conserved bacterial protein involved in outer-membrane-protein assembly. Res Microbiol 155:129–135
Doerrler WT, Raetz CRH (2005) Loss of outer membrane proteins without inhibition of lipid export in an Escherichia coli YaeT mutant. J Biol Chem 280:27679–27687
Werner J, Misra R (2005) YaeT (Omp85) affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol Microbiol 57:1450–1459
Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D (2005) Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245
Tashiro Y, Nomura N, Nakao R, Senpuku H, Kariyama R, Kumon H, Kosono S, Watanabe H, Nakajima T, Uchiyama H (2008) Opr86 is essential for viability and is a potential candidate for a protective antigen against biofilm formation by Pseudomonas aeruginosa. J Bacteriol 190:3969–3978
Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, Silhavy TJ (2007) Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc Natl Acad Sci USA 104:6400–6405
Onufryk C, Crouch ML, Fang FC, Gross CA (2005) Characterization of six lipoproteins in the σE regulon. J Bacteriol 187:4552–4561
Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, Silhavy TJ (2006) YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol 61:151–164
Fussenegger M, Facius D, Meier J, Meyer TF (1996) A novel peptidoglycan-linked lipoprotein (ComL) that functions in natural transformation competence of Neisseria gonorrhoeae. Mol Microbiol 19:1095–1105
Eggert US, Ruiz N, Falcone BV, Branstrom AA, Goldman RC, Silhavy TJ, Kahne D (2001) Genetic basis for activity differences between vancomycin and glycolipid derivatives of vancomycin. Science 294:361–364
Nardini PM, Mellors A, Lo RYC (1998) Characterization of a fourth lipoprotein from Pasteurella haemolytica A1 and its homology to the OmpA family of outer membrane proteins. FEMS Microbiol Lett 165:71–77
Khairnar NP, Kamble VA, Mangoli SH, Apte SK, Misra HS (2007) Involvement of a periplasmic protein kinase in DNA strand break repair and homologous recombination in Escherichia coli. Mol Microbiol 65:294–304
Bredemeier R, Schlegel T, Ertel F, Vojta A, Borissenko L, Bohnsack MT, Groll M, von Haeseler A, Schleiff E (2007) Functional and phylogenetic properties of the pore-forming β-barrel transporters of the Omp85 family. J Biol Chem 282:1882–1890
Nesper J, Brosig A, Ringler P, Patel GJ, Müller SA, Kleinschmidt JH, Boos W, Diederichs K, Welte W (2008) Omp85Tt from Thermus thermophilus HB27: an ancestral type of the Omp85 protein family. J Bacteriol 190:4568–4575
Robert V, Volokhina EB, Senf F, Bos MP, Van Gelder P, Tommassen J (2006) Assembly factor Omp85 recognizes its outer membrane protein substrates by a species-specific C-terminal motif. PLoS Biol 4:1984–1995
Stegmeier JF, Andersen C (2006) Characterization of pores formed by YaeT (Omp85) from Escherichia coli. J Biochem 140:275–283
Struyvé M, Moons M, Tommassen J (1991) Carboxy-terminal phenylalanine is essential for the correct assembly of a bacterial outer membrane protein. J Mol Biol 218:141–148
de Cock H, Struyvé M, Kleerebezem M, van der Krift T, Tommassen J (1997) Role of the carboxy-terminal phenylalanine in the biogenesis of outer membrane protein PhoE of Escherichia coli K-12. J Mol Biol 269:473–478
Sánchez-Pulido L, Devos D, Genevrois S, Vicente M, Valencia A (2003) POTRA: a conserved domain in the FtsQ family and a class of β-barrel outer membrane proteins. Trends Biochem Sci 28:523–526
Bos MP, Robert V, Tommassen J (2007) Functioning of outer membrane protein assembly factor Omp85 requires a single POTRA domain. EMBO Rep 8:1149–1154
Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, Kahne D (2007) Structure and function of an essential component of the outer membrane protein assembly machine. Science 317:961–964
Gatzeva-Topalova PZ, Walton TA, Sousa MC (2008) Crystal structure of YaeT: conformational flexibility and substrate recognition. Structure 16:1873–1881
Knowles TJ, Jeeves M, Bobat S, Dancea F, McClelland D, Palmer T, Overduin M, Henderson IR (2008) Fold and function of polypeptide transport-associated domains responsible for delivering unfolded proteins to membranes. Mol Microbiol 68:1216–1227
Vanini MMT, Spisni A, Sforça ML, Pertinhez TA, Benedetti CE (2008) The solution structure of the outer membrane lipoprotein OmlA from Xanthomonas axonopodis pv. citri reveals a protein fold implicated in protein–protein interaction. Proteins 71:2051–2064
Clantin B, Delattre AS, Rucktooa P, Saint N, Méli AC, Locht C, Jacob-Dubuisson F, Villeret V (2007) Structure of the membrane protein FhaC: a member of the Omp85-TpsB transporter superfamily. Science 317:957–961
Tommassen J (2007) Getting into and through the outer membrane. Science 317:903–904
Wimley WC (2003) The versatile β-barrel membrane protein. Curr Opin Struct Biol 13:404–411
Paschen SA, Neupert W, Rapaport D (2005) Biogenesis of β-barrel membrane proteins of mitochondria. Trends Biochem Sci 30:575–582
Walther DM, Rapaport D (2009) Biogenesis of mitochondrial outer membrane proteins. Biochim Biophys Acta 1793:42–51
Schein SJ, Colombini M, Finkelstein A (1976) Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol 30:99–120
Colombini M (1979) A candidate for the permeability pathway of the outer mitochondrial membrane. Nature 279:643–645
Colombini M (2004) VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem 256–257:107–115
Blachly-Dyson E, Song J, Wolfgang WJ, Colombini M, Forte M (1997) Multicopy suppressors of phenotypes resulting from the absence of yeast VDAC encode a VDAC-like protein. Mol Cell Biol 17:5727–5738
Sampson MJ, Lovell RS, Craigen WJ (1996) Isolation, characterization, and mapping of two mouse mitochondrial voltage-dependent anion channel isoforms. Genomics 33:283–288
Sampson MJ, Lovell RS, Davison DB, Craigen WJ (1996) A novel mouse mitochondrial voltage-dependent anion channel gene localizes to chromosome 8. Genomics 36:192–196
Sampson MJ, Ross L, Decker WK, Craigen WJ (1998) A novel isoform of the mitochondrial outer membrane protein VDAC3 via alternative splicing of a 3-base exon. Functional characteristics and subcellular localization. J Biol Chem 273:30482–30486
Wu S, Sampson MJ, Decker WK, Craigen WJ (1999) Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochim Biophys Acta 1452:68–78
Simamura E, Shimada H, Hatta T, Hirai K (2008) Mitochondrial voltage-dependent anion channels (VDACs) as novel pharmacological targets for anti-cancer agents. J Bioenerg Biomembr 40:213–217
Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399:483–487
Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M (2001) Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem 276:19414–19419
Malia TJ, Wagner G (2007) NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry 46:514–525
Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 9:550–555
Rostovtseva TK, Bezrukov SM (2008) VDAC regulation: role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembr 40:163–170
Baker KP, Schaniel A, Vestweber D, Schatz G (1990) A yeast mitochondrial outer membrane protein essential for protein import and cell viability. Nature 348:605–609
Kiebler M, Pfaller R, Sollner T, Griffiths G, Horstmann H, Pfanner N, Neupert W (1990) Identification of a mitochondrial receptor complex required for recognition and membrane insertion of precursor proteins. Nature 348:610–616
Hill K, Model K, Ryan MT, Dietmeier K, Martin F, Wagner R, Pfanner N (1998) Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins. Nature 395:516–521
Künkele K-P, Heins S, Dembowski M, Nargang FE, Benz R, Thieffry M, Walz J, Lill R, Nussberger S, Neupert W (1998) The preprotein translocation channel of the outer membrane of mitochondria. Cell 93:1009–1019
Kozjak V, Wiedemann N, Milenkovic D, Lohaus C, Meyer HE, Guiard B, Meisinger C, Pfanner N (2003) An essential role of Sam50 in the protein sorting and assembly machinery of the mitochondrial outer membrane. J Biol Chem 278:48520–48523
Gentle I, Gabriel K, Beech P, Waller R, Lithgow T (2004) The Omp85 family of proteins is essential for outer membrane biogenesis in mitochondria and bacteria. J Cell Biol 164:19–24
Paschen SA, Waizenegger T, Stan T, Preuss M, Cyrklaff M, Hell K, Rapaport D, Neupert W (2003) Evolutionary conservation of biogenesis of β-barrel membrane proteins. Nature 426:862–866
Sogo LF, Yaffe MP (1994) Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J Cell Biol 130:1361–1373
Youngman MJ, Hobbs AE, Burgess SM, Srinivasan M, Jensen RE (2004) Mmm2p, a mitochondrial outer membrane protein required for yeast mitochondrial shape and maintenance of mtDNA nucleoids. J Cell Biol 164:677–688
Boldogh IR, Nowakowski DW, Yang HC, Chung H, Karmon S, Royes P, Pon LA (2003) A protein complex containing Mdm10p, Mdm12p, and Mmm1p links mitochondrial membranes and DNA to the cytoskeleton-based segregation machinery. Mol Biol Cell 14:4618–4627
Meisinger C, Rissler M, Chacinska A, Szklarz LK, Milenkovic D, Kozjak V, Schönfisch B, Lohaus C, Meyer HE, Yaffe MP, Guiard B, Wiedemann N, Pfanner N (2004) The mitochondrial morphology protein Mdm10 functions in assembly of the preprotein translocase of the outer membrane. Dev Cell 7:61–71
Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, Vonrhein C, Griesinger C, Zweckstetter M, Zeth K (2008) Structure of the human voltage-dependent anion channel. Proc Natl Acad Sci USA 105:15370–15375
Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G (2008) Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321:1206–1210
Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, Ping P, Abramson J (2008) The crystal structure of mouse VDAC1 at 2.3 Å resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci USA 105:17742–17747
Bosch D, Voorhout W, Tommassen J (1988) Export and localization of N-terminally truncated derivatives of Escherichia coli K-12 outer membrane protein PhoE. J Biol Chem 263:9952–9957
Rapaport D (2003) How to find the right organelle—targeting signals in mitochondrial outer membrane proteins. EMBO Rep 4:948–952
Krimmer T, Rapaport D, Ryan MT, Meisinger C, Kassenbrock CK, Blachly-Dyson E, Forte M, Douglas MG, Neupert W, Nargang FE, Pfanner N (2001) Biogenesis of the major mitochondrial outer membrane protein porin involves a complex import pathway via receptors and the general import pore. J Cell Biol 152:289–300
Model K, Meisinger C, Prinz T, Wiedemann N, Truscott KN, Pfanner N, Ryan MT (2001) Multistep assembly of the protein import channel of the mitochondrial outer membrane. Nat Struct Biol 8:361–370
Rapaport D, Neupert W (1999) Biogenesis of Tom40, core component of the TOM complex of mitochondria. J Cell Biol 146:321–331
Walther DM, Papic D, Bos MP, Tommassen J, Rapaport D (2009) Signals in bacterial β-barrel proteins are functional in eukaryotic cells for targeting to and assembly in mitochondria. Proc Natl Acad Sci USA 106:2531–2536
Court DA, Kleene R, Neupert W, Lill R (1996) Role of the N- and C-termini of porin in import into the outer membrane of Neurospora mitochondria. FEBS Lett 390:73–77
Hamajima S, Sakagushi M, Mihara K, Ono S, Sato R (1988) Both amino- and carboxyterminal portions are required for insertion of yeast porin into the outer mitochondrial membrane. J Biochem (Tokyo) 104:362–367
Müller A, Gunther D, Brinkmann V, Hurwitz R, Meyer TF, Rudel T (2000) Targeting of the pro-apoptotic VDAC-like porin (PorB) of Neisseria gonorrhoeae to mitochondria of infected cells. EMBO J 19:5332–5343
Müller A, Rassow J, Grimm J, Machuy N, Meyer TF, Rudel T (2002) VDAC and the bacterial porin PorB of Neisseria gonorrhoeae share mitochondrial import pathways. EMBO J 21:1916–1929
Kutik S, Stojanovski D, Becker L, Becker T, Meinecke M, Krüger V, Prinz C, Meisinger C, Guiard B, Wagner R, Pfanner N, Wiedemann N (2008) Dissecting membrane insertion of mitochondrial β-barrel proteins. Cell 132:1011–1024
Gratzer S, Lithgow T, Bauer RE, Lamping E, Paltauf F, Kohlwein SD, Haucke V, Junne T, Schatz G, Horst M (1995) Mas37p, a novel receptor subunit for protein import into mitochondria. J Cell Biol 129:25–34
Wiedemann N, Kozjak V, Chacinska A, Schönfish B, Rospert S, Ryan MT, Pfanner N, Meisinger C (2003) Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature 424:565–571
Milenkovic D, Kozjak V, Wiedemann N, Lohaus C, Meyer HE, Guiard B, Pfanner N, Meisinger C (2004) Sam35 of the mitochondrial protein sorting and assembly machinery is a peripheral outer membrane protein essential for cell viability. J Biol Chem 279:22781–22785
Waizenegger T, Habib SJ, Lech M, Mokranjac D, Paschen SA, Hell K, Neupert W, Rapaport D (2004) Tob38, a novel essential component in the biogenesis of β-barrel proteins of mitochondria. EMBO Rep 5:704–709
Ishikawa D, Yamamoto H, Tamura Y, Moritoh K, Endo T (2004) Two novel proteins in the mitochondrial outer membrane mediate β-barrel protein assembly. J Cell Biol 166:621–627
Armstrong LC, Komiya T, Bergman BE, Mihara K, Bornstein P (1997) Metaxin is a component of a preprotein import complex in the outer membrane of the mammalian mitochondrion. J Biol Chem 272:6510–6518
Armstrong LC, Saenz AJ, Bornstein P (1999) Metaxin 1 interacts with metaxin 2, a novel related protein associated with the mammalian mitochondrial outer membrane. J Cell Biochem 74:11–22
Kozjak-Pavlovic V, Ross K, Benlasfer N, Kimmig S, Karlas A, Rudel T (2007) Conserved roles of Sam50 and metaxins in VDAC biogenesis. EMBO Rep 8:576–582
Meisinger C, Pfannschmidt S, Rissler M, Milenkovic D, Becker T, Stojanovski D, Youngman MJ, Jensen RE, Chacinska A, Guiard B, Pfanner N, Wiedemann N (2007) The morphology proteins Mdm12/Mmm1 function in the major β-barrel assembly pathway of mitochondria. EMBO J 26:2229–2239
Habib SJ, Waizenegger T, Niewienda A, Paschen SA, Neupert W, Rapaport D (2007) The N-terminal domain of Tob55 has a receptor-like function in the biogenesis of mitochondrial β-barrel proteins. J Cell Biol 176:77–88
Habib SJ, Waizenegger T, Lech M, Neupert W, Rapaport D (2005) Assembly of the TOB complex of mitochondria. J Biol Chem 280:6434–6440
Schleiff E, Silvius JR, Shore GC (1999) Direct membrane insertion of voltage-dependent anion-selective channel protein catalyzed by mitochondrial Tom20. J Cell Biol 145:973–978
Hoppins SC, Nargang FE (2004) The Tim8–Tim13 complex of Neurospora crassa functions in the assembly of proteins into both mitochondrial membranes. J Biol Chem 279:12396–12405
Wiedemann N, Truscott KN, Pfannschmidt S, Guiard B, Meisinger C, Pfanner N (2004) Biogenesis of the protein import channel Tom40 of the mitochondrial outer membrane: intermembrane space components are involved in an early stage of the assembly pathway. J Biol Chem 279:18188–18194
Smith M, Hicks S, Baker K, McCauley R (1994) Rupture of the mitochondrial outer membrane impairs porin assembly. J Biol Chem 269:28460–28464
Koehler CM (2004) The small Tim proteins and the twin C×3C motif. Trends Biochem Sci 29:1–4
Tokatlidis K, Schatz G (1999) Biogenesis of mitochondrial inner membrane proteins. J Biol Chem 274:35285–35288
Tamm LK, Hong H, Liang B (2004) Folding and assembly of β-barrel membrane proteins. Biochim Biophys Acta 1666:250–263
Xu X, Colombini M (1996) Self-catalyzed insertion of proteins into phospholipid membranes. J Biol Chem 271:23675–23682
Gabriel K, Buchanan SK, Lithgow T (2001) The alpha and beta: protein translocation across mitochondrial and plastid outer membranes. Trends Biochem Sci 26:36–40
Rapaport D (2005) How does the TOM complex mediate insertion of precursor proteins into the mitochondrial outer membrane? J Cell Biol 171:419–423
Chan NC, Lithgow T (2008) The peripheral membrane subunits of the SAM complex function codependently in mitochondrial outer membrane biogenesis. Mol Biol Cell 19:126–136
Schleiff E, Eichacker LA, Eckart K, Becker T, Mirus O, Stahl T, Soll J (2003) Prediction of the plant β-barrel proteome: a case study of the chloroplast outer envelope. Protein Sci 12:748–759
Goetze TA, Philippar K, Ilkavets I, Soll J, Wagner R (2006) OEP37 is a new member of the chloroplast outer membrane ion channels. J Biol Chem 281:17989–17998
Hemmler R, Becker T, Schleiff E, Bölter B, Stahl T, Soll J, Götze TA, Braams S, Wagner R (2006) Molecular properties of Oep21, an ATP-regulated anion-selective solute channel from the outer chloroplast membrane. J Biol Chem 281:12020–12029
Soll J, Schleiff E (2004) Protein import into chloroplasts. Nat Rev Mol Cell Biol 5:198–208
Reumann S, Davila-Aponte J, Keegstra K (1999) The evolutionary origin of the protein-translocating channel of chloroplastic envelope membranes: identification of a cyanobacterial homolog. Proc Natl Acad Sci USA 96:784–789
Eckart K, Eichacker L, Sohrt K, Schleiff E, Heins L, Soll J (2002) A Toc75-like protein import channel is abundant in chloroplasts. EMBO Rep 3:557–562
Patel R, Hsu SC, Bédard J, Inoue K, Jarvis P (2008) The Omp85-related chloroplast outer envelope protein OEP80 is essential for viability in Arabidopsis. Plant Physiol 148:235–245
Schleiff E, Klösgen RB (2001) Without a little help from ‘my’ friends: direct insertion of proteins into chloroplast membranes? Biochim Biophys Acta 1541:22–33
Tranel PJ, Keegstra K (1996) A novel, bipartite transit peptide targets OEP75 to the outer membrane of the chloroplastic envelope. Plant Cell 8:2093–2104
Inoue K, Baldwin AJ, Shipman RL, Matsui K, Theg SM, Ohme-Takagi M (2005) Complete maturation of the plastid protein translocation channel requires a type I signal peptidase. J Cell Biol 171:425–430
Rutten L, Mannie JP, Stead CM, Raetz CR, Reynolds CM, Bonvin AM, Tommassen JP, Egmond MR, Trent MS, Gros P (2009) Active-site architecture and catalytic mechanism of the lipid A deacylase LpxR of Salmonella typhimurium. Proc Natl Acad Sci USA 106:1960–1964
Acknowledgments
We would like to thank Boumediene Soufi for critically reading the manuscript. Our work was supported by grants from the United Mitochondrial Disease Foundation and the Deutsche Forschungsgemeinschaft (SFB 446-A30 and RA 1028/2-1) to D.R., and grants from the Research Council for Chemical Sciences (CW) from the Netherlands Organization for Scientific Research (NWO) to J.T.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Walther, D.M., Rapaport, D. & Tommassen, J. Biogenesis of β-barrel membrane proteins in bacteria and eukaryotes: evolutionary conservation and divergence. Cell. Mol. Life Sci. 66, 2789–2804 (2009). https://doi.org/10.1007/s00018-009-0029-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0029-z