
Abstract
Quantification of the size distributions of organic molecular markers can provide information about the origin of the carbonaceous particulate matter (PM). Organic molecular marker spatial variability studies provide data that are vital to an accurate determination of a population's exposure to PM from various sources. We have investigated the intra-community spatial variation of size-segregated PM [0–0.25 μm (ultrafine), 0.25-2.5 μm (accumulation), and 2.5-10 μm (coarse)) ] in a southern California community. The highest concentrations of individual organic compounds were found in the ultrafine fraction, followed by the accumulation and coarse size fractions. Correlations between the three size fractions were weak between compounds in the coarse and corresponding ultrafine and accumulation particles, implying that the coarse PM organic compounds were emitted by different sources than those that emit ultrafine and accumulation mode PM. Evidence of the incomplete combustion of gasoline was found in the ultrafine and accumulation size fractions, while possible diesel emissions were traced to ultrafine particles. Coefficients of divergence and coefficients of variation were investigated to determine the spatial and temporal variability of individual organic compounds. Spatial divergence in organic compounds was comparatively high, but it did not differ appreciably between size fractions or between compound classes. Elemental carbon and tracer compounds, which originate from a few sources, showed higher spatial divergence than organic carbon whose numerous sources can be local and regional. Spatial and temporal variability were not different from each other for this data set and, therefore, it is not possible to determine whether variability in concentrations between sampling sites or the length of the sampling campaign is more important for health effects studies.
Similar content being viewed by others

Introduction
Exposure to particulate matter (PM) from various sources and in different size fractions has been associated with adverse health impacts (Ritz et al. 2002; Lippmann et al. 2003). Particle size may play a key role in the toxicity of the ambient PM. For example, recent research has shown distinct toxic properties associated with the smallest PM (<0.1 µm) (Li et al. 2003), including exacerbation of asthma (Delfino et al. 2008; Halonen et al. 2008) and cardiovascular effects (Elder et al. 2004). Particulate matter originates directly from multiple primary sources and indirectly by secondary formation mechanisms. It is well known that PM from different sources can have varying degrees of toxicity (Hannigan et al. 2005); therefore, identifying the sources responsible for observed adverse human health will not only provide abatement strategies for the regulation of PM but also insight into the biological mechanisms causing the observed health effects. Determination of the PM source can be achieved through organic speciation analysis, which identifies the chemical composition of the PM and uses this information to discern source origin (Schauer et al. 2002; Querol et al. 2004).
Most of the time series epidemiology studies that have sought to identify relationships between PM levels and human health effects employ one PM sampler in an urban area; thus, a major challenge for improving our understanding of PM and its adverse effects on human health is the necessary improvement in our understanding of the spatial homogeneity of the PM levels at urban or even community scale. Beyond simple PM levels, the spatial distribution of the PM sources must also be assessed (Sheesley et al. 2005; Molitor et al. 2007), as source types such as vehicular emissions are expected to be more locally influenced and can lead to larger exposure characterization errors (Ito et al. 2004). The aim of this study was to determine the spatial variation in size-fractionated organic compounds in a community impacted by numerous sources. Individual organic compounds, specifically the organic molecular markers, were quantified in samples collected concurrently at four sites, located within 2-4 miles of each other. This study provides general trends in molecular marker spatial variability and provides examples of the spatial trends for molecular marker compounds associated with selected PM source categories.
Motor vehicles are significant sources of ambient PM in Southern California (Manchester-Neesvig et al. 2003; Sardar et al. 2005), although other sources, such as wood burning, meat cooking, vegetative detritus, cigarette smoke, and industry, can be important contributors to the overall PM mass (Schauer et al. 1996; Robinson et al. 2006b, c, d). Vehicles emit a variety of organic compounds, but the specific species emitted and their size distribution are related to the fuel type, the engine load conditions, vehicle age and vehicle after-treatment control technology (Riddle et al. 2007b, a; Schauer et al. 2008). Common compound classes of molecular marker compounds emitted by vehicles include alkanes, polycyclic aromatic hydrocarbons (PAHs), and steranes. The PAHs are capable of generating reactive oxygen species and eliciting a state of oxidative stress (Geller et al. 2006), while other compounds act as tracers and are not toxicologically relevant. Identification of specific organic compounds present in vehicular emissions can provide information on the types of fuels burned at the source sites. Tests on gasoline and diesel vehicles have revealed that heavy PAHs are emitted primarily from gasoline vehicles, while light three- and four-ring PAHs are found at very high levels in diesel exhaust (Riddle et al. 2007a, b). Significant spatial and temporal heterogeneity has been found for traffic-related pollutants, such as particle-bound PAHs (Levy et al. 2003).
The degree to which PM chemical components vary on diurnal, seasonal, and annual scales has been investigated (Kim et al. 2002; Fine et al. 2004a, b; Russell et al. 2004; Rinehart et al. 2006; Robinson et al. 2006a; Sheesley et al. 2007), but only limited work has been conducted on the spatial variability of molecular tracer compounds. Within this context, research has focused primarily on differences between source (freeway) and receptor sites (Fine et al. 2004a; Phuleria et al. 2007), highway tunnel emissions in comparison to outdoor ambient concentrations (Fraser et al. 1999), urban area concentrations versus offshore sampling locations (Fraser et al. 2003), differences in organic compound concentrations over large geographical areas (Manchester-Neesvig et al. 2003; Ohura et al. 2004; Zheng et al. 2007), and spatial differences of emissions sources in PM2.5 particles (Rinehart et al. 2006). The study reported here is the first to demonstrate variability in the composition of organic compounds on a community-scale—over an area of 2–4 miles. Others have reported size-fractioned organic concentrations in southern California (Fine et al. 2004a; Miguel et al. 2004; Minguillón et al. 2008; Arhami et al. 2009), but our study is the first to explore the three broad size modes (ultrafine, accumulation, and coarse). By investigating how specific particulate organic molecular markers behave with respect to particle size and spatial distribution, their utility as tracers of primary emissions can be evaluated. In addition, spatial distribution of source contributions will provide vital information for use in source-oriented health models.
Experimental
Ambient sampling
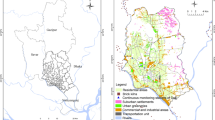
Sampling sites and data collection are described in detail elsewhere (Krudysz et al. 2008). Briefly, the ambient PM samples used in this study were collected by personal cascade impactor samplers (PCIS) on Teflon and quartz-fiber filters over a 10-week period from January to March 2005. The PCIS allows for the collection of size-segregated PM matter in the 0–0.25 μm [quasi-ultrafine; hereafter referred to as ultrafine (UF) for convenience], 0.25–2.5 μm [accumulation (Acc)], and 2.5–10 μm [coarse (C)] size ranges (Singh et al. 2003). A pair of samplers was deployed concurrently at each site for six to seven continuous days (one sampling cycle). At each site, one PCIS was loaded with pre-baked quartz filters for elemental carbon (EC), organic carbon (OC), and individual organic compound analyses, and one was loaded with Teflon filters for particle mass and elemental content determination. Field blanks for both quartz and Teflon filters were also collected at each site and week. The PCIS has been used previously for ambient PM characterization (Majestic et al. 2006). Samples were collected concurrently at four monitoring sites across Long Beach, California (Fig. 1). The sites, north Long beach (NLB), south Long Beach (SLB), Sutter, and Riley are 2–4 miles apart and are impacted by numerous sources, including freeways, ports, airport, power plants, oil refineries, and numerous other local sources.

Sampling locations
Organic speciation
Quantification of individual organic compounds in ambient PM was conducted based on previously developed methods (Mazurek et al. 1987; Schauer et al. 1996; Sheesley et al. 2003). The quartz-fiber filters, including blanks, were spiked with known amounts of isotope-labeled internal standard compounds. Solvent extraction of all samples was conducted according to methods advanced by Fine et al. (2004a) using dichloromethane and methanol. The methods used to analyze and quantify the organic samples were based on those described in Dutton et al. (2008a). In brief, each sample along with a set of authentic quantification standard solutions (PM standards) were analyzed by autoinjection into a gas chromatography–mass spectrometry (GC-MS) system using an Agilent Technologies 6890 N gas chromatograph attached to an Agilent 5975 Inert Mass Selective Detector (MS) (Agilent Technologies, Santa Clara, CA). This system is equipped with a programmable temperature vaporization inlet (Wylie 1997), which enables the user to inject a large volume of solvent into the GC, and with improved detection limits for the organic molecular markers (Crimmins and Baker 2006). Each compound of interest in the sample was referenced to the internal standard that is most similar in chemical structure and size. The peak area ratios of the key ions for the compound of interest and an internal standard with similar structure were calculated. Using a calibration curve, the peak area ratio was then used to calculate the mass ratio for the compound of interest relative to the internal standard. The calibration curve was created using PM standards with compounds of known mass. These PM standards were diluted to five different levels (based on the range of mass expected in the samples) and spiked with the same internal standards used for the samples.
The field blanks were used to determine contamination issues from the filters, handling techniques, and analysis protocols. All measurements for this study were field blank corrected to account for possible bias resulting from background contamination. Many of the field blanks showed levels not significantly different from zero. To reduce sensitivity to occasional outliers in the field blanks, the median rather than the mean value of the ten weekly field blanks was subtracted from all observations.
Uncertainties for each compound and each sample were empirically based on the variance observed in the calibration data (Dutton et al. 2008b), and the root sum of squares (RSS) method (NIST 1994) was used for uncertainty propagation to arrive at the final, blank corrected species concentration uncertainties. Quality control steps were implemented to detect and correct for common problems encountered during organic quantification.
Statistical analysis
This study resulted in a multi-dimensional (time, space, and particle size) data set. As such, four types of relationships were explored:
-
1.
Associations between the same compounds in different size fractions (between-sizes). For example, the correlation between UF-hexacosane, and Acc-hexacosane.
-
2.
Associations between different compounds within each size fraction, (between-compounds). For example, the correlation between UF-hexacosane and UF-benzo(ghi)perylene.
-
3.
Associations between the same compounds from different sites within each size fraction (between-sites). For example, the relationship between NLB-UF-hexacosane and SLB-UF-hexacosane.
-
4.
Comparison between temporal variability by site and its spatial variability by sampling week (temporal–spatial variability). For example, comparison of the variability in all UF-hexacosane measurements across the ten sampling weeks to the variability in UF-hexacosane measurements across the four sites for sampling week one.
Associations between-compounds and between-sizes were determined through Pearson correlation coefficients. The relationship between-sites was determined by calculating coefficients of divergence (COD) for each site pair and organic compound. The COD provides information on the degree of uniformity between sampling sites and is defined as
where x if is the Ith concentration measured at site f, f and h are two different sites, and n is the number of observations. Small COD values imply similarities between sites and, conversely, COD values approaching unity indicate that the sites are very different from each other. Coefficients of divergence have been used previously to determine spatial variability of PM mass and components (Wongphatarakul et al. 1998; Kim et al. 2005; Sardar et al. 2005).
Temporal–spatial variability was assessed by calculating coefficients of variation (CV), defined as the ratio of the standard deviation of all measurements to their mean. Temporal and spatial CVs were calculated to provide the same metric for comparison between spatial and temporal variability. The distribution of COD and CV values are presented as box plots with minimum, maximum, first and third quartiles, and median values.
Results and discussion
Organic compound mass concentrations
Organic compounds selected for analysis in this study were based on their importance in terms of health effects (Mauderly and Chow 2008) and/or utility as markers for source emissions (Schauer et al. 1996). The initial list of organic compounds was further reduced as only compounds present in quantifiable levels in at least one size fraction are discussed here. Quantifiable was operationally defined in our study as a signal-to-noise (S/N) ratio >3. Ultimately, we quantified and analyzed 39 individual organic compounds, including n-alkanes, PAHs, methyl substituted PAHs (methyl-PAHs), steranes, fatty acids, sterols, and methoxyphenols. Tables 1, 2, and 3 show the mean ambient concentrations (ng/m3) and their associated standard deviations calculated for each compound for the UF, Acc, and C size fractions, respectively. These tables also provide the mean quantification uncertainty, S/N, and percentage of below detection limit values (BDL) for each compound and size fraction.
Figure 2 shows size-fractionated time series for selected compounds by sampling site. Organic compound concentrations were generally highest in the UF fraction, followed by Acc and the C size fractions, although concentrations for some compounds at specific sites were comparable between size fractions. In general, concentrations show a small downward temporal trend throughout the sampling period, especially for n-alkanes (docosane through to hentriacontane), and most of the PAHs, but with marginal or no significance. Although the meteorological conditions did not vary much throughout the sampling period (Krudysz et al. 2008), variability in source emission contributions at the sampling sites may have contributed to temporal differences observed in the data. Table S1 in the supplementary materials shows the variability in the week-to-week measurements using CVs for each compound and size fraction. The CVs ranged from 0.23 to 2.28, 0.30 to 2.45, and 0.27 to 3.16 in the UF, Acc, and C size fractions, respectively. The SLB site showed the overall smallest variability in the UF size fraction, with the highest CVs observed in the NLB measurements. The CVs for the Acc and C PM size classes were comparable for all sites.

Size-fractionated time series for selected compounds: a hexacosane, b ab-hopane, c benzo(ghi)perylene. SLB South Long Beach sampling site, NLB north Long Beach sampling site, UF ultrafine particulate matter (PM) size class (diameter 0–0.25 μm), Acc accumulation PM size class (0.25–2.5 μm), C coarse PM size class (2.5–10 μm)
The authors of this study would like to remind the reader that any study of organic aerosols, specifically those using source tracers, need to consider the lifetime of the organic compounds in that specific atmosphere. Recent research has shown that oxidation and other processes can play an important role in organic compound degradation (Robinson et al. 2006a). Photochemistry can have a much larger effect on compounds in the regional air mass than on those emitted locally as the length of time during which processing can occur is much longer. This, however, was not expected to be a major issue during the winter in Los Angeles, because air pollution in the Los Angeles basin is dominated by local emissions mixed with fairly clean background air. Previous studies in the Los Angeles area have shown concentrations (Fine et al. 2004a) comparable with those reported here.
Relationship between size modes (between-sizes)
Correlation analyses can facilitate the identification of compounds originating from similar sources. In addition, the detection of similar compounds in different size fractions can also indicate the processing of particles from smaller to larger sizes. Table 4 shows correlation coefficients (r) across all sites between the three size fractions for each organic compound. Values followed by a superscipt indicate relatively strong correlations (r > 0.70). Only weak correlations were observed between the concentrations in the C fraction and corresponding concentrations of compounds in the UF and Acc fractions, implying that the coarse PM organic compounds quantified here originate from sources that differ from those of the Acc and UF organic compounds. Strong associations were observed between some of the compounds in the UF and the Acc size fractions; for example, n-alkanes (heptacosane through to triacontane, and tritriacontane through to tetratriacontane) show high correlation coefficients between 0.72 and 0.80.
Relatively strong correlations (r = 0.72-0.78, and r = 0.81-0.83) were observed between the UF and Acc low molecular-weight (MW 202–228) and high MW (253–300) PAHs, respectively. These compounds have been associated with gasoline- and diesel-powered motor vehicle emissions (Zielinska et al. 2004; Riddle et al. 2007a, b). Combustion sources are known to emit particles in the UF and Acc size ranges; therefore, the associations between those two size fractions are expected. It is not possible to determine whether similar source contributions or growth of UF particles into Acc aerosols account for stronger associations between the UF and Acc size fractions. Studies have shown, however, that the size distribution of some compounds (steranes and PAHs) emitted by motor vehicles can span both the UF and Acc size fractions investigated in this study (Riddle et al. 2007a, b). Since this is a source intensive area, it is likely that motor vehicle emissions contributed similarly to both size fractions, resulting in high associations.
Emission sources (between-compounds)
Figure 3 shows correlation contour plots for the quantified organic compounds within each size fraction. The differences between the UF and the Acc correlation contour plots can provide information on the differences in the molecular marker composition of sources emitting particles in these size fractions, such as fresh emissions from combustion sources. Strong correlations between the different organic molecular markers within a particle size range may indicate that these compounds originated from similar sources. Overall, the UF and Acc data sets show similarly strong correlations among many of the compounds, although some compounds within the Acc fraction are more highly correlated than those in the UF fraction. The correlations among individual organic compounds in the C fraction are mostly smaller than 0.50. Stronger associations in the Acc size fraction relative to the UF particles imply a stronger influence of local emission sources on UF particles.


Correlation contour plots for compounds within the UF fraction (a), within the Acc fraction (b), and within the C fraction (c)
Concentrations of PAHs ranged from 0.002 to 1.008 ng/m3, which is comparable to those reported in other studies (Fraser et al. 1999; Fine et al. 2004a). Selected PAHs (indeno[1,2,3-cd]pyrene, coronene, benzo[ghi]perylene) have previously been used as molecular markers for motor vehicle gasoline and diesel emissions (Schauer et al. 1999a, 2002; Robinson et al. 2006d; Riddle et al. 2007a, b). Overall, higher PAH levels were observed in the UF fraction relative to the Acc fraction and the C particles, indicative of fresh primary emissions, possibly motor vehicles. Analyses of specific PAHs can provide additional information on the types of vehicular emissions affecting the sampling sites. For example, high correlations between coronene and benzo[ghi]perylene (r = 0.96 and 0.94 for UF and Acc fractions, respectively) have previously been attributed to emissions from poor spark-ignition combustion in gasoline-powered vehicles (Fine et al. 2004a) and incomplete combustion of gasoline deriving from light-duty gasoline vehicles (Kleeman et al. 2008).
Steranes have recently been identified as a reliable marker for motor oil combustion (Kleeman et al. 2008). In this study, steranes were highly correlated with each other in the UF fraction, with slightly lower correlations in the Acc fraction and low associations in the C fraction. Correlation coefficients for steranes ranged from 0.63 (ba-30-norhopane vs. 22 S-ab-30-homohopane) to 0.92 (ba-30-norhopane vs. ab-hopane) for the UF fraction, and 0.65 (ba-30-norhopane vs. 22 S-ab-30-homohopane) to 0.91 (ba-30-norhopane vs. ab-hopane) for the Acc fraction. Very high associations between steranes have been attributed to motor vehicle emissions from urban environments in Pittsburgh (Subramanian et al. 2006) and Los Angeles (Fine et al. 2004a).
In addition to comparing associations within a compound class, we also explored associations across compound classes. In the UF fraction, steranes were not well correlated with n-alkanes and PAHs, while the steranes in the Acc fraction were strongly correlated with the n-alkanes and PAHs. One interpretation of these results is that steranes in the UF fraction are emitted from specific sources that may be different from the sources emitting n-alkanes and PAHs. With respect to Acc particles, similar sources emit steranes, n-alkanes, and PAHs, or the aging and atmospheric transformation of aerosols is responsible for the higher correlations. The contour plots also show that organic compounds quantified in this study were more suitable for identifying sources that emit particles in the UF and Acc size fractions than sources emitting particles in the C fraction.
Steranes and high MW PAHs, such as benzo[ghi]perylene and coronene, have been found in similar concentrations near freeways with gasoline- and diesel-emitting vehicles, while EC and lighter MW PAHs were found at elevated levels near a freeway with more diesel vehicles than near a freeway with only gasoline-emitting vehicles (Phuleria et al. 2007). In the Long Beach area, the highest concentrations of both the low (MW < 228) and high (MW > 252) MW PAHs were found at the Sutter site in the UF and the Acc size fractions. Sutter is the closest site to the I-710 freeway, which has significant (roughly 20%) diesel truck traffic. However, as it is about 650 m from that source, we cannot exclude the influence of other sources of these compounds. No trends were found for the molecular marker composition at the other sampling sites. Although other studies have also found that high- and low MW PAHs are tracers of gasoline-powered and diesel vehicles, respectively (Zielinska et al. 2004), our preliminary analysis of the gasoline and diesel fuel organic markers can not distinguish between the types of vehicular sources most strongly affecting each site in this community scale study.
Ratio–ratio plots can facilitate an investigation of the relative influence of similar sources on the molecular marker concentrations (Robinson et al. 2006b, c, d). The plots compare two species, each normalized by a third mutual species. If there is only one source with consistent emission ratios for all three compounds and if all three compounds are conserved in the atmosphere, then the ambient measurements should cluster around one point on the ratio–ratio plot that corresponds to the emission characteristics of that source. If there are two sources with unique emission rates for the three compounds, the plot will reveal a continuum of points extending along a line between the locations of these two sources on the ratio–ratio plot. If there are three or more sources contributing to the three compounds, the ambient measurements should be constrained to a region defined by the location of the multiple sources on the ratio–ratio plot.
Figure 4 shows a ratio–ratio plot comparing two steranes, ba-30-norhopane and ab-hopane, each normalized by EC. All three size fractions show a continuum of points extending predominantly along a single line, indicating that the compounds were impacted by two sources with unique emission rates. Clear differences are evident between the size fractions, where UF ratios are much lower on the plot compared to Acc and C ratios. Published gasoline and diesel vehicle emission profiles indicate that the points higher on the ratio–ratio plot (corresponding to the Acc and C ratios) originate from gasoline vehicles, while points lower on the ratio–ratio plot (corresponding to the UF ratios) derive from diesel vehicles (Zielinska et al. 1998; Robinson et al. 2006a). The plot shows that the steranes associated with EC appear preferentially in the Acc and C fractions, indicative of motor oil emissions. The EC not associated with motor oil, perhaps diesel vehicle tailpipe emissions, is observed preferentially in the freshly emitted UF particles. This can be visualized by comparing the amount of EC associated with each of the steranes, between the size fractions. The plot shows more EC (higher values in the denominator) in the UF fraction than in the Acc and C particles. This extra EC caused the UF points to be lower down on the continuum in the ratio–ratio plot.

Ratio–ratio plot. EC Elemental carbon
Further evidence that the EC in the UF fraction may be associated with different emission sources than motor oil is illustrated in Fig. 5, which shows box plots of the EC/OC ratios for each site and size fraction. Much higher EC/OC ratios were found in the UF fraction than in the Acc and C size fractions at all sampling sites. Sterane concentrations, however, were comparable in both the Acc and UF particles (Tables 1, 2). Although EC can not be exclusively associated with diesel vehicle emissions (Schauer 2003), the preferential partitioning of EC into the UF size fraction suggests primary emissions other than just motor oil, likely diesel tailpipe emissions.

Box plots (minimum, maximum, first quartile, third quartile, and median) for ratios of EC to organic carbon (OC) by size and site
Spatial variability (between-sites)
Site-to-site variability in individual organic compounds was determined by several methods, including CODs and CVs. Box plots of COD values for each site pair and compound class are shown in Fig. 6. The compound classes were defined such that low and high MW n-alkanes included docosane to tritriacontane and tetratriacontane to octatriacontane, respectively. Low and high MW PAHs included fluoranthene to chrysene/triphenylene and benzo[b&k]fluoranthene to coronene, respectively. The OC and EC COD values are also shown for comparison. Overall, COD values for organic species were found not to differ substantially between size fractions or between compound classes, although high MW n-alkanes showed slightly lower CODs on average in all size fractions. The median COD values for the UF fraction ranged from approximately 0.3 to 0.5, with minimum values similar to the spatial divergence observed for UF OC (COD = approx. 0.18). Similar trends in COD values compared to OC spatial variability were found for the Acc and C size fractions. Numerous local and regional sources contribute to OC concentrations (Schauer et al. 1996), resulting in relatively low spatial divergence, whereas molecular markers are associated with fewer specific sources (Schauer et al. 1999a, b, 2001, 2002); consequently, their spatial distribution is expected to be more variable. The EC COD values were similar or higher than the median COD levels for the organic compounds in all size fractions. Similar to organic marker compounds, EC originated from only a few sources; therefore, the relatively higher spatial variability observed for the organic species results from the variability in specific source emissions. In comparison to the COD values calculated for the inorganic elemental components (Krudysz et al. 2008), the spatial variation in molecular marker compounds is comparably heterogeneous, especially for many of the elemental compounds associated with motor vehicle emissions in the UF and Acc size fractions.

Coefficients of divergence box plots for the UF (a), Acc (b), and C (c) fractions. MW Molecular weight, PAHs polycyclic aromatic hydrocarbons
Temporal–spatial variability
In order to compare spatial to temporal variability within this data set, two sets of CV values were calculated; one for all sites across the sampling period, and the other for each sampling week across all sites. Figure 7 shows the spatial and temporal CVs for each compound class. Box plots represent CV values calculated for each sampling week across the four sites (spatial), while the asterisks represent average values of the four site-specific CVs across all sampling weeks (temporal). Although the spatial CVs show substantial spread, most of the values are very similar to the temporal CVs for this data set. These results imply that longer sampling times and more sampling sites are needed to determine whether variability in concentrations between sampling sites or the length of the sampling campaign is more important for health effects studies.

Spatial (box plots) and temporal (asterisks) coefficients of variation (CV = standard deviation/mean). Spatial CVs were calculated for each sampling week across all sites; temporal CVs are average values of the four site-specific CVs across all sampling weeks
Conclusions
The adverse effects associated with exposure to ambient PM are a significant public health concern. Research must focus on understanding the characteristics of harmful PM and the extent of human exposure to PM. Identification of the sources responsible for the toxic outcomes can enhance their regulation and decrease human exposure. Spatial variability studies are needed to more accurately determine population exposure to PM from various sources. This study presents results from organic speciation analyses from samples collected at four sites of the Long Beach area in southern California in the UF, Acc and C size fractions in a community impacted by numerous sources.
Several degrees of association between the organic compounds, size fractions, and sites were observed in this data set. Across all sites, correlations were stronger for many compounds between UF and Acc size fractions, and weaker between concentrations in the C fraction and corresponding UF and Acc fractions. These results indicate that the coarse PM organic compounds were emitted by sources different than those emitting small particles, such as combustion sources. It is not possible, however, to determine whether different source contributions or atmospheric aerosol processing of smaller particles into larger ones contributed to the similarities in the aerosol chemical makeup.
Associations between the same compounds in different size fractions, across all sites, were performed to gain insight into the types of sources influencing the study sites. Our results show clear differences between size fractions, with stronger correlations within the UF and Acc size fractions compared to those within the C fraction. Differences between correlations within the UF and Acc size fractions, specifically between steranes and n-alkanes, imply that steranes in the UF fraction are emitted from specific sources that may be different from the sources emitting n-alkanes. With respect to the Acc particles, similar sources emit both hopanes and n-alkanes, or aging and atmospheric transformation of aerosols is responsible for the higher correlations. The organic compounds quantified in this study are more suitable for identifying sources that emit particles in the UF and Acc fractions than those that emit particles in the C size fraction.
Strong correlations across all sites were observed between steranes in the UF and the ACC size fractions, and these were attributed to motor vehicle emissions. High correlations between some PAHs in the UF and Acc size fractions were indicative of emissions from poor spark-ignition combustion in gasoline-powered vehicles and incomplete combustion of gasoline derived from light-duty gasoline vehicles.
Ratio–ratio plots were used to investigate the relative influence of similar sources on the molecular marker concentrations. Compounds in all three size fractions were shown to be impacted by two sources with unique emission rates. Steranes associated with EC partition preferentially into the Acc fractions, indicating motor oil emissions as the main PM source, while EC not associated with motor oil, perhaps diesel vehicle tailpipe emissions, is found preferentially in the freshly emitted UF particles. This source hypothesis was further corroborated by the much higher EC/OC ratios in the UF size fraction than in the Acc and C size fractions, whereas steranes were shown to be comparable in both the Acc and UF particles.
Spatial variability was assessed by comparing the same compounds from different sites within each size fraction. The COD values for organic species did not differ appreciably between size fractions or between compound classes, but comparatively high spatial divergence was observed for most species. The molecular tracers were more heterogeneous than OC but comparable to EC. Numerous sources contribute to OC concentrations, resulting in relatively low spatial divergence, whereas EC, like the molecular tracers, originates mostly from traffic sources, which are more local and thus increase its spatial variability. The large variation in spatial distribution of organic compounds and particle sizes presented here (COD = 0.0 – 0.7) suggests that it may be difficult to characterize a community-average concentration for the molecular marker compounds with only one monitoring station or a single particle size. Resolving the spatial variability of PM chemical components will require longer sampling times and a denser network of samplers capable of distinguishing exposure in different microenvironments.
References
Arhami M, Sillanpaa M, Hu S, Olson MR, Schauer JJ, Sioutas C (2009) Size-segregated inorganic and organic components of PM in the communities of the Los Angeles harbor. Aerosol Sci Tech 43(2):145–160. doi:10.1080/02786820802534757
Crimmins BS, Baker JE (2006) Improved GC/MS methods for measuring hourly PAH and nitro-pah concentrations in urban particulate matter. Atmos Environ 40:6764–6779. doi:10.1016/j.atmosenv.2006.05.078
Delfino RJ, Staimer N, Tjoa T, Gillen D, Kleinman MT, Sioutas C, Cooper D (2008) Personal and ambient air pollution exposures and lung function decrements in children with asthma. Environ Health Perspect 116:550–558
Dutton S, Williams D, Garcia J, Vedal S, Hannigan M (2008a) PM2.5 characterization for time series studies: Organic molecular marker speciation methods and observations from daily measurements in Denver. Atmos Environ (submitted)
Dutton SJ, Schauer JJ, Vedal S, Hannigan MP (2008b) PM2.5 characterization in Denver illustrating bulk speciaton and pointwise uncertainty estimation methods for time series studies. Atmos Environ (in press)
Elder A, Gelein R, Finkelstein J, Phipps R, Frampton M, Utell M, Kittelson DB, Watts WF, Hopke P, Jeong CH, Kim E, Liu W, Zhao WX, Zhuo LM, Vincent R, Kumarathasan P, Oberdorster G (2004) On-road exposure to highway aerosols. 2. Exposures of aged, compromised rats. Inhal Toxicol 16:41–53. doi:10.1080/08958370490443222
Fine PM, Chakrabarti B, Krudysz M, Schauer JJ, Sioutas C (2004a) Diurnal variations of individual organic compound constituents of ultrafine and accumulation mode particulate matter in the Los Angeles Basin. Environ Sci Technol 38:1296–1304. doi:10.1021/es0348389
Fine PM, Shen S, Sioutas C (2004b) Inferring the sources of fine and ultrafine particulate matter at downwind receptor sites in the Los Angeles Basin using multiple continuous measurements. Aerosol Sci Tech 38:182–195. doi:10.1080/02786820390229499
Fraser MP, Cass GR, Simoneit BRT (1999) Particulate organic compounds emitted from motor vehicle exhaust and in the urban atmosphere. Atmos Environ 33:2715–2724. doi:10.1016/S1352-2310(98)00311-2
Fraser MP, Cass GR, Simoneit BRT (2003) Air quality model evaluation data for organics. 6. C-3-c-24 organic acids. Environ Sci Technol 37:446–45. doi:10.1021/es0209262
Geller MD, Ntziachristos L, Mamakos A, Samaras Z, Schmitz DA, Froines JR, Sioutas C (2006) Physicochemical and redox characteristics of particulate matter (PM) emitted from gasoline and diesel passenger cars. Atmos Environ 40:6988–7004. doi:10.1016/j.atmosenv.2006.06.018
Halonen JI, Lanki T, Yli-Tuomi T, Kulmala M, Tiittanen P, Pekkanen J (2008) Urban air pollution, and asthma and copd hospital emergency room visits. Thorax 63:635–641. doi:10.1136/thx.2007.091371
Hannigan MP, Busby WF, Cass GR (2005) Source contributions to the mutagenicity of urban particulate air pollution. J Air Waste Manage Assoc 55(4):399–410
Ito K, Xue N, Thurston G (2004) Spatial variation of PM2.5 chemical species and source-apportioned mass concentrations in New York city. Atmos Environ 38:5269–5282. doi:10.1016/j.atmosenv.2004.02.063
Kim E, Hopke PK, Pinto JP, Wilson WE (2005) Spatial variability of fine particle mass, components, and source contributions during the regional air pollution study in St. Louis. Environ Sci Technol 39:4172–4179. doi:10.1021/es049824x
Kim S, Shen S, Sioutas C, Zhu Y, Hinds WC (2002) Size distribution and diurnal and seasonal trends of ultrafine particles in source and receptor sites of the Los Angeles basin. J Air Waste Manage Assoc 52:297–307
Kleeman MJ, Riddle SG, Robert MA, Jakober CA (2008) Lubricating oil and fuel contributions to particulate matter emissions from light-duty gasoline and heavy-duty diesel vehicles. Environ Sci Technol 42:235–242. doi:10.1021/es071054c
Krudysz M, Froines J, Fine P, Sioutas C (2008) Intra-community spatial variation of size-fractionated PM mass, OC, EC, and trace elements in the Long Beach, CA area. Atmos Environ 42(21):5374–5389. doi:10.1016/j.atmosenv.2008.02.060
Levy JI, Bennett DH, Melly SJ, Spengler JD (2003) Influence of traffic patterns on particulate matter and polycyclic aromatic hydrocarbon concentrations in Roxbury, Massachusetts. J Expo Anal Environ Epidemiol 13:364–371. doi:10.1038/sj.jea.7500289
Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A (2003) Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect 111:455–460
Lippmann M, Frampton M, Schwartz J, Dockery D, Schlesinger R, Koutrakis P, Froines J, Nel A, Finkelstein J, Godleski J, Kaufman J, Koenig J, Larson T, Luchtel D, Liu LJS, Oberdorster G, Peters A, Sarnat J, Sioutas C, Suh H, Sullivan J, Utell M, Wichmann E, Zelikoff J (2003) The us environmental protection agency particulate matter health effects research centers program: A midcourse report of status, progress, and plans. Environ Health Perspect 111:1074–1092
Majestic BJ, Schauer JJ, Shafer MM, Turner JR, Fine PM, Singh M, Sioutas C (2006) Development of a wet-chemical method for the speciation of iron in atmospheric aerosols. Environ Sci Technol 40:2346–2351. doi:10.1021/es052023p
Manchester-Neesvig JB, Schauer JJ, Cass GR (2003) The distribution of particle-phase organic compounds in the atmosphere and their use for source apportionment during the Southern California children's health study. J Air Waste Manage Assoc 53:1065–1079
Mauderly JL, Chow JC (2008) Health effects of organic aerosols. Inhal Toxicol 20:257–288. doi:10.1080/08958370701866008
Mazurek MA, Simoneit BRT, Cass GR, Gray HA (1987) Quantitative high-resolution gas-chromatography and high-resolution gas-chromatography mass-spectrometry analyses of carbonaceous fine aerosol-particles. Int J Environ Ch 29:119–139. doi:10.1080/03067318708078415
Miguel AH, Eiguren-Fernandez A, Jaques PA, Froines JR, Grant BL, Mayo PR, Sioutas C (2004) Seasonal variation of the particle size distribution of polycyclic aromatic hydrocarbons and of major aerosol species in Claremont, California. Atmos Environ 38:3241–3251. doi:10.1016/j.atmosenv.2004.03.008
Minguillón MC, Arhami M, Schauer JJ, Olson MR, Sioutas C (2008) Seasonal and spatial variations of sources of fine and quasi-ultrafine particulate matter in neighborhoods near the Los Angeles-long beach harbor. Atmos Environ 42(32):7317–7328. doi:10.1016/j.atmosenv.2008.07.036
Molitor J, Jerrett M, Chang CC, Molitor NT, Gauderman J, Berhane K, McConnell R, Lurmann F, Wu J, Winer A, Thomas D (2007) Assessing uncertainty in spatial exposure models for air pollution health effects assessment. Environ Health Perspect 115:1147–1153
National Institute of Standards and Technology (NIST) (1994) Technical note 1297, 1994 edition: guidelines for evaluating and expressing the uncertainty of nist measurement results. NIST, Gaithersburg. Available at: http://physics.Nist.Gov/pubs/guidelines/appa.Html
Ohura T, Amagai T, Fusaya M, Matsushita H (2004) Spatial distributions and profiles of atmospheric polycyclic aromatic hydrocarbons in two industrial cities in japan. Environ Sci Technol 38:49–55. doi:10.1021/es030541z
Phuleria HC, Sheesley RJ, Schauer JJ, Fine PM, Sioutas C (2007) Roadside measurements of size-segregated particulate organic compounds near gasoline and diesel-dominated freeways in Los Angeles, CA. Atmos Environ 41:4653–4671. doi:10.1016/j.atmosenv.2007.03.031
Querol X, Alastuey A, Ruiz CR, Artinano B, Hansson HC, Harrison RM, Buringh E, ten Brink HM, Lutz M, Bruckmann P, Straehl P, Schneider J (2004) Speciation and origin of PM10 and PM2.5 in selected European cities. Atmos Environ 38:6547–6555. doi:10.1016/j.atmosenv.2004.08.037
Riddle SG, Robert MA, Jakober CA, Hannigan MP, Kleeman MJ (2007a) Size distribution of trace organic species emitted from heavy-duty diesel vehicles. Environ Sci Technol 41:1962–1969. doi:10.1021/es0617183
Riddle SG, Robert MA, Jakober CA, Hannigan MP, Kleeman MJ (2007b) Size distribution of trace organic species emitted from light-duty gasoline vehicles. Environ Sci Technol 41:7464–7471. doi:10.1021/es070153n
Rinehart LR, Fujita EM, Chow JC, Magliano K, Zielinska B (2006) Spatial distribution of PM2.5 associated organic compounds in central California. Atmos Environ 40:290–303
Ritz B, Yu F, Fruin S, Chapa G, Shaw GM, Harris JA (2002) Ambient air pollution and risk of birth defects in southern California. Am J Epidemiol 155:17–25. doi:10.1093/aje/155.1.17
Robinson AL, Donahue NM, Rogge WF (2006a) Photochemical oxidation and changes in molecular composition of organic aerosol in the regional context, J Geophys Res 111. doi: 10.1029/.2005/JD006265
Robinson AL, Subramanian R, Donahue NM, Bernardo-Bricker A, Rogge WF (2006b) Source apportionment of molecular markers and organic aerosols-1. Polycyclic aromatic hydrocarbons and methodology for data visualization. Environ Sci Technol 40:7803–7810. doi:10.1021/es0510414
Robinson AL, Subramanian R, Donahue NM, Bernardo-Bricker A, Rogge WF (2006c) Source apportionment of molecular markers and organic aerosol. 3. Food cooking emissions. Environ Sci Technol 40:7820–7827. doi:10.1021/es060781p
Robinson AL, Subramanian R, Donahue NM, Bernardo-Bricker A, Rogge WF (2006d) Source apportionment of molecular markers and organic aerosol. 2. Biomass smoke. Environ Sci Technol 40:7811–7819. doi:10.1021/es060782h
Russell M, Allen DT, Collins DR, Fraser MP (2004) Daily, seasonal, and spatial trends in PM2.5 mass and composition in southeast Texas. Aerosol Sci Technol 38:14–26. doi:10.1080/02786820390229318
Sardar SB, Fine PM, Sioutas C (2005) Seasonal and spatial variability of the size-resolved chemical composition of particulate matter (PM10) in the Los Angeles basin. J Geophys Res 110. doi:10.1029/2004JD004627
Schauer JJ (2003) Evaluation of elemental carbon as a marker for diesel particulate matter. J Expo Anal Environ Epidemiol 13:443–453. doi:10.1038/sj.jea.7500298
Schauer JJ, Rogge WF, Hildemann LM, Mazurek MA, Cass GR, Simoneit BRT (1996) Source apportionment of airborne particulate matter using organic compounds as tracers. Atmos Environ 30:3837–3855. doi:10.1016/1352-2310(96)00085-4
Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT (1999a) Measurement of emissions from air pollution sources. 2. C-1 through c-30 organic compounds from medium duty diesel trucks. Environ Sci Technol 33:1578–1587. doi:10.1021/es980081n
Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT (1999b) Measurement of emissions from air pollution sources. 1. C-1 through c-29 organic compounds from meat charbroiling. Environ Sci Technol 33:1566–1577. doi:10.1021/es980076j
Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT (2001) Measurement of emissions from air pollution sources. 3. C-1-c-29 organic compounds from fireplace combustion of wood. Environ Sci Technol 35:1716–1728. doi:10.1021/es001331e
Schauer JJ, Kleeman MJ, Cass GR, Simoneit BR (2002) Measurement of emissions from air pollution sources. 5. C1-c32 organic compounds from gasoline-powered motor vehicles. Environ Sci Technol 36:1169–1180. doi:10.1021/es0108077
Schauer JJ, Christensen CG, Kittelson DB, Johnson JP, Watts WF (2008) Impact of ambient temperatures and driving conditions on the chemical composition of particulate matter emissions from non-smoking gasoline-powered motor vehicles. Aerosol Sci Technol 42:210–223. doi:10.1080/02786820801958742
Sheesley RJ, Schauer JJ, Chowdhury Z, Cass GR, Simoneit BRT (2003) Characterization of organic aerosols emitted from the combustion of biomass indigenous to south Asia. J Geophys Res 108 (D9). doi:10.1029/2002JD002981
Sheesley RJ, Schauer JJ, Hemming JD, Geis S, Barman MA (2005) Seasonal and spatial relationship of chemistry and toxicity in atmospheric particulate matter using aquatic bioassays. Environ Sci Technol 39:999–1010. doi:10.1021/es049873+
Sheesley RJ, Schauer JJ, Meiritz M, DeMinter JT, Bae MS, Turner JR (2007) Daily variation in particle-phase source tracers in an urban atmosphere. Aerosol Sci Technol 41:981–993. doi:10.1080/02786820701644277
Singh M, Misra C, Sioutas C (2003) Field evaluation of a personal cascade impactor sampler (PCIS). Atmos Environ 37:4781–4793. doi:10.1016/j.atmosenv.2003.08.013
Subramanian R, Donahue NM, Bernardo-Bricker A, Rogge WF, Robinson AL (2006) Contribution of motor vehicle emissions to organic carbon and fine particle mass in Pittsburgh, Pennsylvania: Effects of varying source profiles and seasonal trends in ambient marker concentrations. Atmos Environ 40:8002–8019. doi:10.1016/j.atmosenv.2006.06.055
Wongphatarakul V, Friedlander SK, Pinto JP (1998) A comparative study of PM2.5 ambient aerosol chemical databases. Environ Sci Technol 32:3926–3934. doi:10.1021/es9800582
Wylie PL (1997) Trace level pesticide analysis by GC/MS using large-volume injection. Application Note: gas chromatography. Agilent Technologies, Wilmington, pp 1-10
Zheng M, Cass GR, Ke L, Wang F, Schauer JJ, Edgerton ES, Russell AG (2007) Source apportionment of daily fine particulate matter at Jefferson street, Atlanta, GA, during summer and winter. J Air Waste Manage Assoc 57:228–242
Zielinska B, McDonald J, Hayes T, Chow JC, Fujita EM, Watson JG (1998) Northern front range air quality study final report, volume B: Source measurements. Prepared for Colorado State University, Fort Collins, Colorado. Desert Research Institute, Reno
Zielinska B, Sagebiel J, Arnott WP, Rogers CF, Kelly KE, Wagner DA, Lighty JS, Sarofim AF, Palmer G (2004) Phase and size distribution of polycyclic aromatic hydrocarbons in diesel and gasoline vehicle emissions. Environ Sci Technol 38:2557–2567. doi:10.1021/es030518d
Acknowledgments
The research described in this paper was supported by the Center for Occupational and Environmental Health, the National Institute of Environmental Health Sciences, the Southern California Particle Center (SCPC), funded by EPA under the STAR program through Grant RD-8324-1301-0, the Southern California Environmental Health Sciences Center (SCEHSC) grant number 5P30 ES07048, and in part by the United States Environmental Protection Agency (EPA) under the Science to Achieve Results (STAR) fellowship program and a STAR grant number R832157. The EPA has not officially endorsed this publication and the views expressed herein may not reflect the views of the EPA.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
Mean value of the field blank corrections expressed as a concentration and coefficient of variation (CV = standard deviation/mean) values for each site, compound, and size fraction (DOC 36 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Krudysz, M.A., Dutton, S.J., Brinkman, G.L. et al. Intra-community spatial variation of size-fractionated organic compounds in Long Beach, California. Air Qual Atmos Health 2, 69–88 (2009). https://doi.org/10.1007/s11869-009-0035-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11869-009-0035-1