
Abstract
The spike (S) protein is a key structural protein of coronaviruses including, the porcine transmissible gastroenteritis virus (TGEV). The S protein is a type I membrane glycoprotein located in the viral envelope and is responsible for mediating the binding of viral particles to specific cell receptors and therefore specific cell types. It is also an important immune target for the host in neutralizing the virus. Four antigenic sites A, B, C, and D that reside near the N-terminal domain have been defined in the S protein. Of these, the region encoding antigenic sites A and to a lesser extent D, herein defined as S-AD, are most critical in eliciting host neutralizing antibodies. Herein, we enzymatically amplified, cloned, and expressed the S-AD fragment from TGEV in the prokaryotic expression vector, pET-30a. Maximum protein expression was achieved at 30°C over a 5-h period post-induction. Rabbit polyclonal antiserum was generated using recombinant S-AD (rS-AD) protein. In contrast to prior studies showing no activity with bacterially produced S protein, results indicated that polyclonal serum recognized TGEV-infected cells and reduced infection by 100%. Furthermore, the truncated rS-AD peptide was able to bind to the surface of cells from swine testes in a competitive manner and completely inhibit viral infection.
Similar content being viewed by others


Introduction
Transmissible gastroenteritis virus (TGEV) belongs to the family Coronaviridae. It is the etiological agent of transmissible gastroenteritis (TGE). Although all age groups of pigs are susceptible to TGEV infection, the mortality rate can reach as high as 100% in seronegative suckling piglets. TGEV is a pleomorphic enveloped virus with a positive-stranded RNA genome that consists of four structural proteins: the spike (S) protein, the integral membrane (M) protein, the minor envelope (E) protein, and the nucleocapsid (N) protein [1]. Among these, the surface protein S, also an integral membrane protein, is the major host target for generating neutralizing antibodies to the virus. The binding of the S protein to the cellular receptor, porcine aminopeptidase N (pAPN), mediates the attachment of viruses to host cells before cell invasion [2]. The TGEV S gene encodes a 1,447-amino acid precursor polypeptide containing a 16-residue signal peptide and a hydrophobic region near the C-terminus that maybe involved in anchoring the S protein to the viral envelope [3].
There are four antigenic sites (in the order of C, B, D, and A) in the amino terminal half of the S protein [4]. Site A is the major inducer of neutralizing antibodies [5] and highly conserved in TGEV and Porcine Respiratory Coronavirus (PRCV), a respiratory variant of TGEV [6]. Deletion of S gene residues encoded by bases 621–681 leads to a loss of one or two antigenic sites (C and B or D) depending on the nomenclature [5].
In this study, we expressed a truncated TGEV S protein containing both the A and D antigenic sites by cloning this region into a bacterial expression system. We then ascertained the ability of rabbit polyclonal anti-serum and/or the recombinant protein (rS-AD) to inhibit infection in vitro. Our results indicate that the rS-AD not only induces specific antibodies recognizing TGEV-infected eukaryotic cells, but can interfere with cell infection by the virus. In addition, rabbit polyclonal antibodies to rS-AD were capable of neutralizing TGEV when the virus was pretreated with the immune serum.
Materials and methods
Construction of expression plasmid, pET-S-AD
Forward (PETTGS1: 5′-GGGCGAATTCATGACTCTTGAAATTTCATGTTAT) and reverse (PETTGS2: 5′-CCGGGTCGACTTTTATAACAGCTGTGGCATCTAA) primers were used to amplify antigenic sites A and D (bases 1102–1812) in a single fragment from a recombinant plasmid pCG1-TGEVS containing the full-length TGEV S gene (kindly provided by Drs. G. Herrler and C. Schwegmann-Wessels) [7]. The relative positions of the four antigenic sites in the S protein of TGEV and location of the cloned gene bearing antigenic sites A and D are shown in Fig. 1. The primers contained EcoRI and SalI sites (underlined), respectively, to facilitate cloning. The PCR reaction (25 μl) consisted of 1 μg template, 50 pmol of each primer, 200 μM dNTPs, 1.25 U ExTaq polymerase (TaKaRa, China), and 1× PCR buffer. The PCR involved a preincubation step at 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 55.9°C for 30 s, and 72°C for 1 min; an extension at 72°C for 10 min was included at the end of the cycling. The purified PCR product was cloned into the EcoRI:SalI site of pET-30a (Novagen, USA). The resulting recombinant plasmid, designated pET-S-AD, was purified with a commercially available kit (KeyGen Biotech, China) and verified by sequencing.

Schematic representation of the four antigenic sites (A–D) in the S protein of TGEV. The antigenic sites (in order of C, B, D, and A) of the TGEV S protein using the Madrid nomenclature are shown. The relative positions and the regions spanned by each antigenic site are indicated by black boxes. The location of the rS-AD gene is also provided
Expression and purification of recombinant protein
The plasmid pET-S-AD was transformed into host cells, Escherichia coli BL21(DE3) pLysS for protein expression. Protein production was optimized according to Liu et al. [8] with minor modification. In brief, bacteria harboring pET-S-AD were cultured in LB at 37°C and induced with 1 mM isopropyl β-d-thiogalactoside (IPTG, Sigma, China) when the OD600 reached 0.5. Induction took place at 30°C for 5 h. Bacteria transformed with empty pET-30a vector were similarly treated for use as a negative control. After the induction period, all bacteria were pelleted, re-suspended in TE buffer (50 mM Tris and 1 mM EDTA, pH 8.0), and treated with lysozyme (100 μg/l) at room temperature (RT) for 30 min. The cells were washed with PBS then sonicated on ice for 30 min. After centrifugation at 10,000 rpm for 20 min, aliquots of the supernatants and pellets were subjected to 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The gel was scanned and the amount of protein approximated using Glyko BandScan software (v 5.0) according to the manufacturer’s instructions.
Purification of inclusion bodies and renaturation of the fusion protein by dialysis were performed as described by Liu et al. [8]. In brief, bacterial pellets re-suspended in buffer II (50 mM Tris, 0.5 mM EDTA, and 1% Triton-100, pH 8.0) were centrifuged at 10,000 rpm for 10 min at 4°C. The pelleted inclusion bodies were washed with PBS and re-suspended in SDS-PAGE loading buffer, boiled for 10 min, and centrifuged at 12,000 rpm for 5 min. The resulting supernatant was subjected to 12% SDS-PAGE, and the sample-containing gel was washed with non-ionized water. Regions with high protein content were visualized by immersing the gel in 0.25 M cold KCl dissolved in PBS for 30 min. The protein of interest was excised from the gel and homogenized in PBS. After triple freezing and thawing with liquid nitrogen, the samples were centrifuged at 5,000 rpm for 10 min, and the supernatant containing purified protein was designated rS-AD.
Preparation and titration of rabbit polyclonal antiserum
To obtain polyclonal antibodies, a New Zealand rabbit was subcutaneously injected with 1 ml of purified rS-AD protein (2 mg/ml) emulsified with Freund’s complete adjuvant. After 2 weeks and for 6 consecutive weeks thereafter, the rabbit was boosted with the same antigen emulsified with Freund’s incomplete adjuvant. Control serum was generated by immunizing a second rabbit with adjuvants only. Animals were bled through the ear vein 1 week following the final injection.
Antiserum titers were evaluated by indirect ELISA. In brief, ELISA plates were coated with rS-AD protein using 10, 1, or 0.1 μg per well (100 μl/well) at 4°C, overnight in carbonate–bicarbonate buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 8.6). The next day, the wells were incubated with blocking buffer (5% non-fat dry milk and 0.05% Tween-20 in PBS) for 1 h, rinsed 2× with PBS-0.05% Tween 20 (PBST), then incubated with serially diluted rS-AD antibody for 1 h. Serum from the sham-immunized rabbit was used as a control in parallel wells. Peroxidase-conjugated goat anti-rabbit IgG was used as the second antibody followed by o-phenylenediamine (OPD) substrate (Sigma, China) for color development. The procedure was performed at 37°C and the reaction terminated with 2 M H2SO4. Absorbance values were read at OD490 using an ELISA reader.
Western blots
Purified rS-AD, unfused vector protein (negative control) and TGEV particles (positive control) were subjected to SDS-PAGE and transferred to nitrocellulose (NC). Rabbit antiserum to rS-AD was diluted 1:1,000 in PBS and incubated at RT for 2 h with the NC membranes precut into strips. Peroxidase-labeled goat anti-rabbit IgG (Boster, China) was diluted 1:2,000 and incubated for 1 h with the PBST-washed NC to detect the rS-AD or viral S proteins, respectively. The protein bands were visualized using OPD.
Polyclonal antibody recognition of TGEV-infected cells
Swine testis (ST) cells, a transformed cell line, were cultured in Eagle’s minimal essential medium (EMEM) containing 5% newborn bovine serum (ExCell Biol. Inc, Shanghai, China). The cells were seeded onto coverslips in 24-well plates, washed three times with PBS, then infected with TGEV strain Purdue 46-MAD (provided by Dr. Luis Enjuanes; CSIC-UAM Canto Blanco, Madrid, Spain) at a titer of 104pfu/ml. A conventional indirect immunofluorescence assay was performed at 24-h post-infection using the rabbit anti-(rS-AD) serum as the primary antibody (1:1000 dilution). Fluoresceine isothiocyanate-labeled goat anti-rabbit IgG was used as the secondary antibody, and green fluorescence signals were recorded by fluorescence microscopy (Leica, Germany).
Inhibition of TGEV binding to ST cells
The maximum nontoxic concentration of rS-AD that could be applied to ST cells was determined using cell viability as a read out. Trypan blue staining and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (Sigma, China) cell proliferation assays were performed in 96-well plates according to published protocols [9, 10] using serially diluted rS-AD protein. The rS-AD (25 μg/ml) was then used at the maximum nontoxic concentration and at serially diluted concentrations to test the ability of rS-AD protein to inhibit infection by TGEV in vitro. In brief, the ST cells were first seeded on to 24-well plates at 37°C and allowed to adhere for 24–36 h. The cells were washed 3× with PBS, then incubated for 2 h with serially diluted rS-AD or with similar concentrations of bacterially expressed S protein from avian infectious bronchitis virus (IBV) (negative control) which was purified using the same procedure. After incubation, the cells were again washed 3× with PBS to remove residual protein. Antigen-treated ST cells were incubated with varying concentrations of TGEV at multiplicities of infection (MOI) of 0.1, 1, or 10 for 1 h, washed 3× with PBS, and then incubated for an additional 36 h to allow sufficient time for internalization and viral replication before performing virus plaque reduction assays.
In parallel, various concentrations of the rS-AD rabbit antiserum were pre-incubated with TGEV at 37°C. After 1 h, ST cells previously seeded in 24-well plates were incubated with antiserum-treated viruses at a titer of 104pfu/ml at 37°C for approximately 1 h. Virus plaque reduction assays were performed 36 h later following extensive washing of the cells. Serum from the sham-immunized rabbit served as a negative control. All cultured ST cells were subjected to virus plaque reduction assays using crystal violet staining [9, 11] to quantify the responses. All the experiments using rS-AD and polyclonal antiserum were performed in triplicate.
RT-PCR
To further analyze the inhibitory effects of anti-SAD antibody on cell infection, a RT-PCR was performed. In brief, TGEV of 100TCID50 (50% tissue culture infective dose) was incubated with serially diluted anti-SAD antibody for 1 h at 37°C then used to infect ST cells on 6-well plates at 37°C for 1 h. After washing extensively in PBS, the cells were cultured with serum-free DMEM. The non-antibody-treated TGEV-infected and mock-infected ST cells were used as controls. After 2 days, the cells were subjected to triple freezing and thawing followed by pelleting and resuspension in sterile water. The resulting suspension was subjected to RNA extraction using a commercially available kit (Qiagen, Germany) according to the manufacturer’s instructions. qRT-PCR was used to amplify a small portion of the 3′-region of the S gene, an intervening sequence, and the 3a gene (designated S-X; 192 bp) using sense 5′-GGCTTAGTAGTAATATTTTGCATAC and anti-sense 5′-ATTATAGCAGATGATAGAATTAACA primers. The housekeeping gene beta-actin (208 bp) was also amplified by qRT-PCR as an internal reference using sense and antisense primers 5′-GGCTCAGAGCAAGAGAGGTATCC and 5′-GGTCTCAAACATGATCTGAGTCATCT, respectively [12]. All amplifications were performed using a RT-PCR kit (TaKaRa, Japan) according to the manufacturer’s instructions. The PCR profile for amplification of TGEV-S-X gene began with 95°C for 10 min followed by 30 cycles of 94°C for l min, 48°C for 1 min, and 72°C for l min. There was a final extension at 72°C for l0 min. Amplification of beta-actin was performed as described for the S-X gene except that the annealing temperature was 58.8°C. The PCR products were separated on a 1% agarose gel. Quantification of the gene amplification was performed using the gel documentation system (Uvitec, Cambridge, UK) and determined with Gel Analyzer software (Copyright 2010 by Dr. Istvan Lazar) according to the manufacturer’s instructions [13].
Statistical analysis
Comparisons between two different groups were determined with the Student’s t test. Multiple comparisons were conducted using one-way ANOVA. All statistical analyses were performed using SPSS 16.0 software where P < 0.01 was considered statistically significant. All data were represented as the mean ± SEM.
Results
Sequencing of the cloned S-AD insert indicated that the PCR product was 710 bp in length and corresponded to bases 1102–1812 of the TGEV S gene (GenBank accession No. M94101). The amount of protein expression accounted for approximately 35% of the total bacterial protein produced (Fig. 2a) as estimated from scanned SDS-PAGE gels. The rS-AD fusion protein was mainly expressed within inclusion bodies in the bacterial cytosol; the fusion protein was approximately 34 kDa in size.

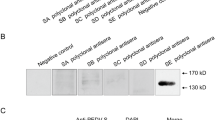
Expression and detection of rS-AD. Expression of rS-AD was monitored by a Coomassie blue staining of PAGE gels and b Western blot analysis of PAGE gels screened with rabbit antiserum (1:1,000 dilution) to rS-AD as the primary antibody. a M protein markers (kDa). Lane 1 crude bacterial lysate from cells containing non-recombinant vector only; Lane 2 crude bacterial lysate from cells containing the rS-AD plasmid; Lane 3 purified rS-AD protein derived from lysate in lane 2. b Lane 1 purified proteins from bacteria producing rS-AD; Lane 2 non-recombinant vector protein; Lane 3: homogenate from purified and concentrated TGEV. The size of both proteins is indicated
Western blots showed that rS-AD was recognized specifically by rabbit anti-(rS-AD) serum. This same serum recognized the full-length, native S protein of TGEV (Fig. 2b). In indirect immunofluorescence assays, green fluorescence was detected on the surface of the TGEV-infected ST cells using the polyclonal antibody produced in this study (Fig. 3a). No fluorescence signal was found when the mock-infected cells were tested (Fig. 3b) or when negative control rabbit serum was used (data not shown).

Immunofluorescence staining of TGEV-infected ST cells. Porcine ST cells were grown in the presence (TGEV infection) or absence (mock infection) of TGEV (104 pfu/ml). After extensive washing, the cells were incubated with rabbit antiserum to rS-AD (1:1000 dilution) followed by fluoresceine isothiocyanate-labeled goat anti-rabbit IgG secondary antibody (1:2,000)
The maximum nontoxic concentration of the protein was empirically determined to be 25 μg/ml. At this concentration, the cells were not stained by trypan blue, and the high cell survival rate was confirmed by MTT. Pre-incubation of ST cells with this concentration of rS-AD resulted in complete inhibition of cell infection by TGEV; inhibition occurred in a dose-dependent manner and was first observed with as little as 1.56 μg/ml rS-AD (Fig. 4a). When rabbit anti-(rS-AD) antibodies were incubated with TGEV before exposing the virus to ST cells, a dose-dependent and remarkable reduction in infection was also observed (Fig. 4b). The inhibitory effects of the anti-(rS-AD) antibodies on cell infection by TGEV were further investigated by RT-PCR. The results showed that when viral particles pretreated with increasing amounts of antibody were used to infect ST cells, the amount of viral RNA extracted and amplified from cell lysates decreased in a dose-dependent manner as the amount of antibody used to pretreat the virus increased showing nearly complete inhibition at a 1/16 dilution of antibody (Fig. 5).

Inhibition of TGEV infection of ST cells using rS-AD (a) or rS-AD rabbit anti-rS-AD (b). a ST cells were pre-treated with serially diluted rS-AD (maximum 25 μg/ml) or control protein (IBV S; maximum 25 μg/ml). Cells were then incubated with TGEV at various MOIs as shown. b TGEV at a final concentration of 104 pfu/ml was pre-incubated with serially diluted rabbit anti-(rS-AD) or with similarly diluted serum from a sham-immunized rabbit (control) then used to infect ST cells. In both A and B, viral inhibition was monitored by virus plaque reduction assays. Data are presented as averages of three independent trials and are statistically significant (P < 0.01). Error bars represent standard deviation

RT-PCR amplification of the TGEV-S-X gene. Total RNA was extracted from TGEV-infected cells (Virus), non-infected cells (Cell), or cells infected with TGEV pretreated with varying concentrations of anti-(rS-AD) before infection. The qRT-PCR data based upon the TGEV-S-X site was normalized to that of beta-actin. Serum dilutions are given on the x axis. Results are averages of no less than three independent experiments. A representative agarose gel for visualization of the RT-PCR product is provided
Discussion
Among the key structural proteins of TGEV, the S protein has received particular attention because it is associated with several important biological functions of the virus such as pathogenicity and cell fusion. Most importantly, the host immune system efficiently recognizes this S protein and produces neutralizing antibodies capable of interfering with recognition and infection of the cell by the virus. Four antigenic sites (A, B, C, and D) have been defined by competition among monoclonal antibodies that target these sections of the protein. Prior study has shown that antigenic sites A and D are predominantly responsible for inducing neutralizing host antibodies [14]. The full-length S protein was previously expressed using E. coli [15], vaccinia virus [16], and adenovirus [17]; however, TGEV-neutralizing antibodies were only elicited with protein generated from recombinant virus. Inasmuch as the S protein is a glycoprotein, the lack of glycosylation in the prokaryotic expression system may have accounted for the absence of neutralizing antibodies. Nevertheless, prokaryotic expression systems have advantages in protein production, manipulation, convenience, and production costs [18]. Heterologous proteins have been expressed successfully in prokaryotic vectors for generating antibodies, neutralizing viruses, or analyzing protein function [19–21].
It has been documented that antigenic site A is fully dependent on glycosylation for proper folding and site D is only partially independent on glycosylation [13]. In our study, we chose to focus only on those sites within the S protein responsible for inducing neutralizing antibodies in the hope of obviating some of the short comings from working with the full-length protein in a prokaryotic system. As expected, our cloned gene fragment generated a high level of the rS-AD protein. This peptide was tested both as an inhibitor of virus infectivity by preventing binding to the cell as well as an immunogen to generate rabbit polyclonal antibodies for obviating viral attachment to ST cells. The antibodies did indeed recognize TGEV-infected cells, indicating that antigenic sites other than those dependent upon glycosylation are present within the A and D regions. In addition, we confirmed that the polyclonal antibodies reacted with transiently expressed full-length S protein via Western Blots. The possibility of using this protein to develop specific monoclonal antibodies is under investigation.
Inhibitory effects of the rS-AD protein on TGEV infection were examined by pre-incubating the recombinant protein with ST cells before introducing the virus so as to competitively inhibit the virus from fusing to the cell. Plaque reduction assays clearly showed that inhibition of viral infection approached 100% relative to negative controls. Recently, we also demonstrated that pre-incubation of recombinant S1 protein from avian bronchitis virus with TGEV was unable to inhibit cell infection by TGEV [22] offering further evidence that the phenomenon observed here was specific. As a note added in proof, we also tested the antibodies against other strains of the virus with similar levels of success (data not shown). In the presumed absence of glycosylation, our data suggest that the primary and/or secondary sequence of the truncated rS-AD are capable of inhibiting cell binding to the A and D regions of the viral S protein when cells are pretreated with recombinant protein. It is interesting to consider whether or not other truncated constructs involving A, B, C, and/or D might have similar blocking effects observed here. Inasmuch as antigenic sites A and D are proximally positioned within the N-terminal region of the S protein, it is possible that the rS-AD may also contain one or more important receptor binding domains.
In our study, rabbit anti-(rS-AD) also completely inhibited cell infection by TGEV. Our results clearly showed that while the rabbit anti-(rS-AD) may be useful as a diagnostic reagent to detect TGEV by ELISA, Western blots or immunofluorescence, the antibody is capable of completely neutralizing TGEV in vitro at the higher concentrations used in our studies. To better characterize the binding of rS-AD, we hope to investigate the interaction between the recombinant construct produced here and aminopeptidase N, the cellular receptor for TGEV.
References
W. Spaan, D. Cavanagh, C. Horzinek, J. Gen. Virol. 69, 2939–2952 (1988)
B. Delmas, J. Gelfi, R. L’Haridon, L.K. Vogel, H. Sjostrom, O. Noren, H. Laude, Nature 357, 417–420 (1992)
M. Godet, J. Grosclaude, B. Delmas, H. Laude, J. Virol. 68, 8008–8016 (1994)
L. Enjuanes, C. Suñé, F. Gebauer, C. Smerdou, A. Camacho, I.M. Antón, S. González, A. Talamillo, A. Méndez, M.L. Ballesteros et al., Vet. Microbiol. 33, 249–262 (1992)
C.M. Sanchez, F. Gebauer, C. Sune, A. Mendez, J. Dopazo, L. Enjuanes, Virology 190, 92–105 (1992)
B. Delmas, D. Rasschaert, M. Godet, J. Gelfi, H. Laude, J. Gen. Virol. 71, 1313–1323 (1990)
C. Schwegmann-Wessels, J. Glende, X. Ren, X. Qu, H. Deng, L. Enjuanes, G. Herrler, J. Gen. Virol. 90, 1724–1729 (2009)
B. Liu, G. Li, X. Sui, J. Yin, H. Wang, X. Ren, J. Biotechnol. 141, 91–96 (2009)
J. Li, J. Yin, X. Sui, G. Li, X. Ren, Avian Pathol. 38, 1–7 (2009)
X. Sui, J. Yin, X. Ren, Antiviral Res. 85, 346–353 (2010)
X. Ren, J. Glende, J. Yin, C. Schwegmann-Wessels, G. Herrler, Virus Res. 137, 220–224 (2008)
X. Ren, F. Meng, J. Yin, G. Li, X. Li, C. Wang, G. Herrler, PLoS One 6, e18669 (2011)
X. Ren, P. Li, Virus Genes 42, 229–235 (2011)
F. Gebauer, W.P. Posthumus, I. Correa, C. Suñé, C. Smerdou, C.M. Sánchez, J.A. Lenstra, R.H. Meloen, L. Enjuanes, Virology 183, 225–238 (1991)
S. Hu, J. Bruszewski, T. Boone, L. Souza, in Modern Approaches to Vaccines, ed. by R.M. Chanock, R.A. Lerner (Cold Spring Harbor Laboratory, New York, 1984), pp. 219–223
D.J. Pulford, P. Britton, Virology 182, 765–773 (1991)
J.M. Torres, C. Sánchez, C. Suñé, C. Smerdou, L. Prevec, F. Graham, L. Enjuanes, Virology 213, 503–516 (1995)
J. Yin, G. Li, X. Ren, G. Herrler, J. Biotechnol. 127, 335–347 (2007)
X. Ren, M. Wang, J. Yin, G. Li, J. Clin. Microbiol. 48, 1875–1881 (2010)
X. Ren, M. Wang, J. Yin, Y. Ren, G. Li, J. Biotechnol. 147, 130–135 (2010)
X. Ren, B. Liu, J. Yin, H. Zhang, G. Li, Virology 410, 299–306 (2011)
X. Ren, G. Li, B. Liu, J. Biotechnol. 150, 202–206 (2010)
Acknowledgments
We thank Dr. Luis Enjuanes, CSIC, Madrid, Spain, for providing TGEV and Drs. Georg Herrler and Christel Schwegmann-Wessels, University of Veterinary Medicine Hannover, Germany, for providing the S gene. This study is funded in part by the National Natural Science Foundation of China (30972195). Additional funding is supported by the Open Project Program of Beijing Key Laboratory of Traditional Chinese Veterinary Medicine at Beijing University of Agriculture (TCVM-201103); the Program for New Century Excellent Talents at the Heilongjiang Provincial University (1155–NCET–005) and supported by the New Century Excellent Talents program from the Ministry of Education of P. R. China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Meng, F., Zhao, Z., Li, G. et al. Bacterial expression of antigenic sites A and D in the spike protein of transmissible gastroenteritis virus and evaluation of their inhibitory effects on viral infection. Virus Genes 43, 335–341 (2011). https://doi.org/10.1007/s11262-011-0637-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-011-0637-1