
Abstract
T-DNA insertion mutants have been widely used to investigate plant gene functions. Unexpectedly, in several reported cases, the phenotype of T-DNA insertion mutations can be suppressed because of trans T-DNA interactions associated with epigenetic modification, which indicates that caution is needed when T-DNA mutants are used. In the present study, we characterized a novel process suppressing a T-DNA mutation. The spz2 (suppressor of zou 2) mutant was isolated as a suppressor of the phenotype of the zou-4 mutant caused by a T-DNA insertion in the first intron. The spz2 mutation partially recovered the native ZOU gene expression in the zou-4 background, but not in two other zou alleles, zou-2 and zou-3, with T-DNAs inserted in the exon and intron, respectively. The suppressed phenotype was inherited in a Mendelian fashion and is not associated with epigenetic modification. The recovery of the native ZOU gene expression in the spz2 zou-4 double mutant is caused by transcriptional read-through of the intronic T-DNA as a result of decreased proximal polyadenylation. SPZ2 encodes an RNA-binding protein, FPA, which is known to regulate polyadenylation site selection. This is the first example of FPA rescuing a T-DNA insertion mutation by affecting the polyadenylation site selection.
Similar content being viewed by others


Introduction
Transfer DNA (T-DNA) insertion is a highly effective mutagen for genome-wide mutagenesis and has been widely used by the Arabidopsis research community to elucidate individual gene function as well as interactions among gene networks (Krysan et al. 1999; Alonso et al. 2003). The insertion of T-DNAs into any part of a gene leads to a diverse range of outcomes. T-DNA inserted in an exon may create null alleles because of the introduction of a premature stop codon or a frame-shift mutation, whereas a T-DNA inserted in an intron may generate null alleles because of failure of proper splicing, leading to an aberrant/truncated protein (Wang 2008), or because of transcriptional termination within the T-DNA sequence. These intronic insertions presumably utilize the endogenous plant splicing and/or polyadenylation complexes; however, until now no such factors have been shown to influence the phenotypes of intronic T-DNA mutations.
The effect of intronic T-DNA insertions upon gene activity can be alleviated by splicing out the T-DNA together with the surrounding intron, and this is not an unusual phenomenon (Ulker et al. 2008; Wang 2008). Recently, RNA/RNA interaction has been implicated in modifying the consequences of intronic T-DNA insertions. This interaction may involve epigenetic modification triggered by the trans-interaction of different T-DNA insertions. For example, the mutant phenotype of a SALK T-DNA insertion in the first intron of the COBRA gene was suppressed by crossing with another T-DNA mutant srf6-1 or other randomly selected SALK T-DNA lines (Xue et al. 2012). In this case, the suppression was associated with increased DNA methylation, which somehow resulted in increased expression of the wild-type COBRA transcript. Another example concerns the AG-TD allele containing a T-DNA insertion in the second intron of the AGAMOUS gene, which is suppressed by other mutants that contain the same T-DNA sequence, such as yuc1-1 (Gao and Zhao 2013). Again, the suppression is inherited in a non-Mendelian (epigenetic) fashion and is associated with silencing of a kanamycin resistance gene located in the T-DNA, suggesting that DNA methylation may be involved. The brassinosteroid-catabolism mutation ben1-1 is transformed to a partial loss-of-function mutation in the bas1-2 sob7-1 ben1-1 (triple-mutant) background and shows enhanced levels of the wild-type-spliced transcript (Sandhu et al. 2013). The instability of the intronic T-DNA mutation csn5a-2 is caused by epigenetic modification when another T-DNA mutation, rop11-1, is introduced (Jia et al. 2015).
ZOU/RETARDED GROWTH OF EMBRYO1 (ZOU/RGE1) plays a key role in endosperm cell death (Kondou et al. 2008; Yang et al. 2008) and embryonic cuticle formation during Arabidopsis seed development. The endosperm surrounds the embryos of angiosperm seeds and is the second zygotic product of double fertilization (Olsen 2004). The endosperm cells breakdown in the embryo surrounding region (ESR) in both monocotyledons and dicotyledons, thus freeing the nutrients that fuel the embryo and make space for the expanding embryo (Ingram 2010). The ZOU gene encodes a helix-loop-helix transcription factor exclusively expressed in the ESR endosperm. The heterodimerization of ZOU and INDUCER OF CBF EXPRESSION1 (ICE1)—another bHLH transcription factor with broader, well-characterized roles in regulating tolerance to cold stress (Chinnusamy et al. 2003; Lee et al. 2005; Miura et al. 2007) and stomatal development (Kanaoka et al. 2008)—is strictly necessary for ZOU target gene activation (Denay et al. 2014). The zou/rge1 mutants have lower ESR cell death compared to the wild type, and the endosperm persists in the mature seeds. The ABNORMAL LEAF SHAPE1 (ALE1) gene is a target of ZOU and is expressed in the ESR and required for normal embryo cuticle formation (Tanaka et al. 2001). Transgene complementation studies suggest that the downregulated expression of ALE1 that occurs in the zou mutant is largely responsible for the embryo cuticle defect but not for endosperm cell death. Endosperm breakdown and the ALE1 regulated cuticle formation are thus two genetically separable pathways regulated by ZOU (Xing et al. 2013).
FPA, a spen family protein with three repeated RNA recognition motifs (RRMs), was first identified as a flowering time regulator in Arabidopsis. Mechanistically, FPA is a trans-acting regulator of poly(A) site selection that participates in alternative cleavage and polyadenylation. Its mutation can bring about intergenic read-through and chimeric RNA formation due to selection of alternative polyadenylation and RNA 3′-end sites (Hornyik et al. 2010; Duc et al. 2013). FPA promotes flowering by downregulating expression of the floral repressor FLC. The basis for this is complex, but involves FPA regulating the relative levels of different antisense transcripts from the FLC locus through its effects on RNA 3′-end formation. FPA was also implicated in RNA silencing by RNA-dependent DNA methylation on account of fpa mutations suppressing RNAi-directed transcriptional gene silencing (Schomburg et al. 2001; Bäurle et al. 2007). However, the DNA methylation and direct RNA sequencing analysis in fpa-7 show no evidence of FPA affecting RNA-dependent DNA methylation, and it is now thought that the effects on siRNA silenced targets may be a consequence of transcript read-through from adjacent loci (Stroud et al. 2013; Duc et al. 2013). Here, we show that fpa mutations suppress the mutant seed phenotype of a specific zou mutant allele containing an intronic T-DNA insertion. This is the first example showing that disruption of gene function by an intronic T-DNA insertion can be dependent on the proximal polyadenylation activity provided by the FPA gene. Our work may also provide a new perspective in understanding the alternative polyadenylation in plants.
Results
Identification of the suppressors of zou mutant
The zou-4 mutant with T-DNA inserted in the ZOU first intron confers the characteristic zou “shrivelled seed phenotype” (Fig. 1b). The seeds of this mutant have a non-degrading endosperm, and the embryo is arrested at the heart-torpedo stage (Fig. 1e). In order to isolate suppressors of the zou-4 mutation (which might help identify other genes involved in endosperm breakdown), we mutagenized zou-4 plants using ethyl methanesulfonate (EMS) or random T-DNA insertion. In total, 23 suppressor mutants were identified from the 2 mutagenesis populations, 16 from the EMS treatment and the remaining 7 from T-DNA mutagenesis. All of these suppressors showed rounder and bigger seeds than the zou-4 mutant. The suppressor mutants were named according to the order in which they were found, as suppressor of zou 1-23 (spz1-23). As all of them are from the zou-4 mutant in the Col-0 background, we labeled them as spz zou-4 in this article. When spz zou-4 mutants were backcrossed reciprocally to the zou-4 progenitor, the resulting F1 and F2 populations indicated that each spz mutation affected a single nuclear gene.

The spz2 mutation suppresses the zou shrinking seed phenotype. a Seeds from Col-0 wild type. b Shrivelled seeds from the zou-4 mutant. c Seeds from the spz2 zou-4 homozygous mutant, which suppressed the zou-4 mutant phenotype, comprised both normal-looking and misshapen seeds. d Paraffin section of Col-0 wild-type seed at the mature stage. e Paraffin section of zou-4 mutant seed with persistent endosperm and heart stage embryo in mature seeds. f Paraffin section of spz2 zou-4 mature seed with fully expanded embryo and one layer of persistent endosperm (the aleurone, also found in wild-type seed). Scale bars 1.0 mm in A–C, and 100 μm in D–F
The suppressor spz2 zou-4 with round seeds and late flowering phenotype was selected for investigation. Although the mature seeds in spz2 zou-4 were rounder and bigger compared to the zou mutant seeds, no more than 20 % of the mature seeds were normal like wild type, and the rest showed defects in seed shape (Table 1; Fig. 1a, c). The embryo could expand to later stages than heart or torpedo, and the endosperm showed breakdown in spz2 zou-4 mature seeds as in wild type (Fig. 1d, f). We obtained shrivelled F1 seeds from the cross between the spz2 zou-4 homozygotic line and the zou-4 mutant. The shrivelled F1 seeds were similar to those of the zou mutant (both maternal and paternal crossings). The F2 seeds originating from self-pollinated F1 plants segregated with a 3:1 ratio of shrivelled and rounded seeds (χ2 = 0.07, P = 0.79 > 0.5; Table 2). This demonstrated that the spz2 mutant was a single recessive mutant with normal transmission through male and female gametophytes. Moreover, spz2 acts zygotically rather than maternally to control seed development.
Molecular cloning and characterization of SPZ2 gene
In spz2 cloning, the positional region was restricted to 75 kb on chromosome 2: 17,980,415–18,055,543 bp, which had 29 genes (Fig. 2a). The genomic sequencing of this region showed that a C to T transition in the 2473 bp of the At2g43410 coding region mutated the amino acid Gln825 to a stop codon (Fig. 2d). This nonsense mutation caused the premature termination of translation in the spz2 mutant. The At2g43410 gene codes the FPA gene, which plays a role in the regulation of flowering time by affecting the FLC mRNA level in the autonomous flowering pathway (Hornyik et al. 2010).

Cloning and characterization of SPZ2 and its mutant alleles. a Map-based cloning of spz2. b The independently isolated spz10 allele also suppressed the zou-4 mutant seed phenotype. c The independently isolated spz23 allele also suppressed the zou-4 mutant seed phenotype. d The genomic structure of SPZ2/FPA, the lesions present in spz2, spz23 and the position of the T-DNA inserted at spz10 are shown. Scale bars 1.0 mm in B and C
To confirm the cloning result, a complementation assay was performed by introducing a wild-type FPA gene copy into the spz2 zou-4 background by floral dip transformation. The seed phenotype of spz2 zou-4 was complemented with full and shrivelled seed phenotypes segregating in the T2 seed produced on primary (T1) transformant plants (Supplementary Fig. S1b, c). The T1 generations of spz2 zou-4 transgenic lines also showed complementation for the late flowering phenotype (Supplementary Fig. S1a). The complementation confirmed that the mutation in FPA was responsible for the suppression of the zou mutant phenotype in the spz2 zou-4 background.
To determine whether other spz mutations were also alleles of FPA, we crossed the spz2 zou-4 line with the other 22 spz zou-4 mutants. The F1 and F2 seeds from the reciprocal crosses with the two spz mutants, spz10 zou-4 and spz23 zou-4, were round, indicating that spz10 and spz23 were also fpa alleles (Supplementary Fig. S2a, b; Table 1). The spz10 zou-4 mutant was obtained from the T-DNA insertion mutagenesis population (Fig. 2b) and spz23 zou-4 from the EMS mutagenesis population (Fig. 2c). Furthermore, the homozygous lines of these three mutants and their hybrid progenies showed a delayed flowering phenotype (Supplementary Table 1). Molecular analysis further revealed that the spz23 mutant harbored a C to T transition at position 2122 in the FPA coding sequence (with +1 indicating the A of the ATG start codon) resulting in the mutation of Gln708 to a stop codon (Fig. 2d; Supplementary Fig. S2c). The spz10 mutant has a T-DNA (pSKI074) insertion in the first intron (1508 bp downstream of ATG) and a 30-bp deletion at the site of T-DNA insertion (Fig. 2d; Supplementary Fig. S2c). We further confirmed the genotype of the F1 generations in allelic analysis among spz2, spz10 and spz23 and found that the mutant sites of the three alleles were heterozygous in the F1 generation (Supplementary Fig. S2d–g). These results confirmed that spz2, spz10 and spz23 were allelic mutants of the FPA gene.
In order to determine the FPA expression profile during seed development, we constructed GUS reporter lines. Histochemical staining of the reporter lines for GUS gene activity on different days after germination showed that the FPA was expressed throughout the plant life cycle especially in young and actively growing tissues such as root tips and leaf margins (Supplementary Fig. S3a–c), which is consistent with the results of Schomburg et al. (2001). Our results also showed that the GUS signal was evident in both the endosperm and the embryo throughout seed development (Supplementary Fig. S3d–f).
The zou phenotype suppression by spz2 is dependent on the zou-4 background
The spz2 mutation was a recessive mutation in a single gene as the F2 population from spz2 zou-4 backcrossed to zou-4 segregated 3:1 for shrivelled:round seeds. However, in the spz2 mapping population derived from spz2 zou-4 (Col-0 background) crossed with another zou allele—zou-2 (Ws-0 background), the shrivelled and rounded seed segregation ratio was 4.3:1 (13:3), which is higher than the 3:1 ratio of a single recessive mutation (Table 2). This indicated the spz2 suppression phenotype was dependent on the genetic background (the 13:3 ratio is as predicted if zou-2/zou-2 homozygotes are not suppressed by spz2). In order to determine the genetic background effects on seed phenotype, we genotyped 50 individuals of the mapping F2 population. We found four spz2 zou-2 homozygous individuals with shrivelled seed phenotypes similar to the zou mutant. A further experiment showed the progeny of these four spz2 zou-2 homozygotes also had 100 % shriveled seeds. This result showed that the spz2 mutation cannot restore the zou-2 shrinking seed phenotype, suggesting that the rescue of the zou phenotype by spz2 mutation may be specific for the zou-4 allele.
To verify the genetic background effect, spz2 zou-4/Col-0 was crossed to zou-3, which was also in the Col-0 ecotype with a T-DNA inserted in the first intron of the ZOU gene (Supplementary Fig. S4). Among the 324 F2 seeds, there were 262 shrinking and 62 rounded seeds with a segregation ratio 4.3:1 as in the previous spz2 zou-4/Col-0 crossed with the zou-2/Ws-0 F2 population (Table 2). After genotyping of 80 individuals from this F2 population, we found 6 spz2 zou-3 homozygous plants with shrinking phenotype similar to the zou mutant. This result indicated that the SPZ2 mutation also cannot restore the zou shrinking seed phenotype in the zou-3/Col-0 background. This confirmed that the spz2 suppressed zou phenotype depends on the zou-4 background and is not associated with the plant ecotype in Col-0 or Ws-0. Moreover, the allelic lines of spz2, spz10 and spz23 suppressed the zou shrinking seed phenotype only in the zou-4 mutation background and not in the zou-2 and zou-3 background (data not shown). Therefore, we conclude that the SPZ2 mutation only restores the zou mutation phenotype in the zou-4 background.
spz2 suppresses the zou-4 phenotype because of restoring the full-length ZOU mRNA
To determine the reason for spz2 suppressing the zou-4 phenotype, we characterized the ZOU transcripts in both the zou-4 single mutant and the spz2 zou-4 double mutant. Our results showed that the full-length ZOU transcript could be amplified in the spz2 zou-4 double mutant, but not in zou-4 (Fig. 3a). Sequencing of this ZOU cDNA from spz2 zou-4 showed that the recovered ZOU transcript was identical to the wild type. We further compared the expression of wild-type ZOU in spz2 zou-4 with Col-0 and zou-4 using qPCR. The expression of the full-length ZOU mRNA was about ten-fold lower in spz2 zou-4 seed than in wild type (Fig. 3c). Consistently, the read density map of ZOU from RNA seq profiles also demonstrated that exons 2 and 3 of ZOU after the T-DNA insertion site were recovered in spz2 zou-4, but not in zou-4 (Fig. 3d). Moreover, we did not detect the full-length ZOU transcript in the spz2 zou-3 homozygotes consistent with their non-rescued seed phenotype (Fig. 3b). Thus, the suppression of the zou mutant phenotype in zou-4 can be attributed to the restoration of full-length ZOU mRNA expression in spz2 zou-4.

Full-length native ZOU transcript is detected in spz2 zou-4. a The full-length coding region of the ZOU transcript was amplified. At 35 cycles, the ZOU transcript was detectable in spz2 zou-4 but not in zou-4. b The full-length coding region of the ZOU transcript was amplified compared with spz2 zou-4 (positive control) and zou-4 (negative control). At 35 cycles, the ZOU transcript was detectable in spz2 zou-4 but not in spz2 zou-3 and zou-4. c qRT-PCR analysis of ZOU expression in the wild type, zou-4 and spz2 zou-4 double mutant. d Read density map of ZOU visualized with the Integrative Genomics Viewer (IGV). Exons 2 and 3 were not expressed in zou-4 but were recovered in spz2 zou-4 mutant
The intronic T-DNA suppression in spz2 zou-4 was not caused by epigenetic modification
Although suppression of the mutant phenotypes caused by intronic T-DNA insertions has been described previously in a few cases, most of the characterized cases were associated with trans T-DNA-mediated epigenetic modifications. In trans T-DNA-mediated intronic T-DNA suppression, the rescued phenotype persists following re-isolation of the intronic T-DNA single mutant from the double or triple mutants and is inherited in a non-Mendelian fashion; also it often shows loss of the antibiotic resistance conferred by the selectable marker gene in the T-DNA, presumably due to DNA methylation and gene silencing (Xue et al. 2012; Gao and Zhao 2013; Sandhu et al. 2013; Jia et al. 2015). Given that spz2 is not a T-DNA induced mutation, this explanation seems unlikely in our case. However, to further test whether the suppression is from epigenetic modifications, we characterized the genetic behavior of the spz2 zou-4 mutant. The F1 generation seeds from crossing spz2 zou-4 with SPZ2 zou-4 all showed the shrinking seed phenotype (Table 2), i.e., they behaved as a stable recessive mutation unlike the above examples involving epigenetic modification (in several of the other examples, the suppressed T-DNA allele is paramutagenic and can induce suppression in a naïve, non-suppressed allele). Further genotyping of the 531 F2 generation seeds of spz2 zou-4 with SPZ2 zou-4 showed that all 137 rounded-seed plants are spz2 homozygotes, and the remaining 394 shrinking-seed plants are SPZ2/spz2 heterozygotes or SPZ2/SPZ2 homozygotes (Table 2). This result demonstrated that the restored zou-4 phenotype occurs only in the spz2 mutated homozygous seeds and the rescued phenotype followed classical Mendelian inheritance, unlike the other cases in which once triggered, the suppressed epialleles were maintained independently of the T-DNA mutant triggering the suppression.
To check the activity of the sulfadiazine resistance gene harbored by the T-DNA in zou-4, we assayed sulfadiazine resistance in zou-4 and spz2 zou-4 seedlings. Both were resistant to sulfadiazine (Fig. 4a), and the transcripts of the sulfonamide resistance gene were accumulated threefold higher in spz2 zou-4 than in zou-4 (Fig. 4b).

The suppression was not correlated with the loss of the antibiotic resistance. a The sulfadiazine resistance trait, conferred by the T-DNA resistance marker gene in zou-4, was sustained in the spz2 zou-4 double mutant. Seedlings were grown on plant media with sulfadiazine (left) and without (right) for 7 days before photographing. b The expression of the sulfadiazine resistance gene in zou-4 and spz2 zou-4 detected by qPCR
The restored expression of native ZOU gene in spz2 zou-4 was associated with alternative polyadenylation
As shown above, in the reads density map of the ZOU gene from gene expression profiles, exons 2 and 3 downstream of the T-DNA insertion site were transcribed in spz2 zou-4 but not in zou-4 (Fig. 3d). This indicated that the spz2 mutation could promote the expression of the native ZOU gene beyond the T-DNA insertion site. To further confirm this result, we analyzed the transcription products transcribed from the ZOU transcription start site in both zou-4 and spz2 zou-4 by 3′ RACE (Fig. 5). The RACE experiment showed that the wild-type ZOU transcripts comprised two alternatively spliced transcripts (α and β: 1054 and 939 bp in length, respectively) in Col-0 and were also detected in spz2 zou-4, whereas neither was detected in zou-4 (Fig. 5b). On the contrary, three truncated transcripts were detected in the zou-4 mutant: transcript 1 contained the first exon and terminated in the ZOU first intron; transcript 2 and 3 comprised ZOU exon 1 and a part of the T-DNA sequence with alternatively spliced and polyadenylation sites (Fig. 5a, b, d). In the spz2 zou-4 mutant, all five transcripts were identified including the two wild-type ZOU transcripts (α and β) and three chimeric transcripts detected in zou-4 (Fig. 5b). The chimeric RNA expression in zou-4 and spz2 zou-4 detected by qPCR showed that all chimeric transcripts in spz2 zou-4 were decreased in comparison with zou-4. Transcript 1 was decreased one fold in spz2 zou-4 compared with zou-4 and transcript 2 plus transcript 3 in spz2 zou-4 was decreased five fold compared with zou-4 (Fig. 5c). Consequently, chimeric transcripts were generated in zou-4 by transcription terminating in the proximal polyadenylation sites, whereas the termination efficiency in these proximal polyadenylation sites was decreased, resulting in read-through of the complete ZOU genomic DNA and splicing out of the T-DNA with an intron in the spz2 zou-4 double mutant. This demonstrated that these chimeric transcripts in zou-4 were dependent on FPA activity to terminate their transcription by polyadenylation, and the recovery of full-length ZOU expression in spz2 zou-4 was a result of transcriptional read-through with reduced premature transcription termination.

Expression of the chimeric transcripts produced by the intronic T-DNA insertion at zou-4 that inactivates the ZOU gene is reduced in spz2 zou-4. a Schematic representation of the gene structure of ZOU in the zou-4 mutant and the alternative polyadenylation products. b 3′ RACE product detected by southern blot with a probe corresponding to the exon 1 region of ZOU. c qRT-PCR analysis of the chimeric transcript expression in zou-4 and spz2 zou-4. The abundances of the chimeric transcripts were all decreased in spz2 zou-4 compared with zou-4. d Sequencing of the chimeric transcripts amplified in zou-4 and spz2 zou-4 by RT-PCR. The chimeric transcripts were comprised of transcripts with differing alternative polyadenylation and alternative splicing sites
SPZ2 plays a wide role in seed development
In order to characterize the role of FPA in seed development, we performed transcriptional profiling (by RNA seq) of Col-0, zou-4 and spz2 zou-4 seed (at the heart stage of embryo development). Comparison of Col-0 and zou-4 seed should identify ZOU targets. Comparison of spz2 zou-4 and zou-4 seeds will identify targets of FPA but also ZOU target genes whose expression is rescued by the partial restoration of ZOU expression in spz2 zou-4. In the comparison of spz2 zou-4 and Col-0 seeds, FPA targets will be identified but also ZOU target genes whose expression is not fully rescued by the partial restoration of ZOU expression in spz2 zou-4. Genes that are both differentially expressed in spz2 zou-4 compared with Col-0 and zou-4 and dismiss the differentially expressed in Col-0 compared with zou-4 are therefore likely to be enriched for genes regulated directly or indirectly by FPA. However, the limitation of the above comparisons is that it is difficult to exclude the zou mutant effects in identifying SPZ2 regulated genes.
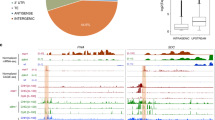
According to the above comparisons, we identified 609 common differentially expressed genes caused by the SPZ2 mutation (Fig. 6a; Supplementary Table 2). Among the 609 genes, 446 were increased and 163 were decreased in spz2. The differentially expressed genes were functionally annotated based on the gene ontology (GO; Fig. 6b). The GO category analysis revealed that the up- and downregulated genes mostly fell into the same categories. In the cellular component, most of the genes were located in the cell part and organelle. In the biological process, most of the genes were involved in the metabolic process, cellular process, biological regulation and response to stimulus. In molecular function, the main GO categories were catalytic activity and binding activity. There were only five genes belonging to the RNA-binding category, namely AT5G18180, RNS3, CID9, AT4G30800 and PUM9. AT5G18180 is involved in rRNA processing, and RNS3 is a ribonuclease protein. CID9 is a putative RNA-binding protein. AT4G30800 is a ribosomal protein, and PUM9 is involved in seed dormancy by regulating both mRNA stability and translation through sequence-specific binding to the 3′ UTR of target mRNA transcripts (Xiang et al. 2014). Unexpectedly, no genes were identified to associate with the RNA polyadenylation process.

Identification and characterization of the differentially expressed genes caused by spz2 mutation. a Comparison of differentially expressed genes among Col-0_Vs_zou-4, spz2 zou-4_Vs_zou-4 and spz2 zou-4_Vs_Col-0. Red box represents the differentially expressed genes caused by spz2 mutation. b GO annotation of the differentially expressed genes due to spz2 mutation. c Interaction networks of the differentially expressed genes in cluster I. The FPA protein is represented by the red box. The lines of different colors represent the types of evidence upon which the associations are based: green neighborhood evidence, red gene fusion evidence, blue co-occurrence evidence, black coexpression evidence, purple experimental evidence, cyan database evidence, yellow textmining evidence, light blue homology evidence
The interactions among differentially expressed genes were analyzed by the STRING database (Supplementary Fig. S5). There were 220 genes connected in the interaction network among the 609 differentially expressed genes. The FPA closely interacted with flower regulator FLC, acetyl-CoA carboxylase ACC2 and RNA-binding protein AT5G18180 (Fig. 6c). Besides this, three large interactive clusters were in this network. The first one is based on acetyl-CoA carboxylase (ACC2), protein phosphatase 2C family protein (At3g51370) and highly ABA-induced PP2C gene 3 (HAI3) related protein kinase genes and cytochrome P450 genes interactions (Fig. 6c); the second one is based on the ABI five binding protein 3 (AFP3) and ABA-responsive element-binding protein (ABF3) correlated stress tolerance network; the third one is exopolygalacturonase (At3g14040), putative pectinesterase (At5g07410) and pectin methylesterases such as the protein (VGD1) organized cell wall modification network. The GO analysis and interaction network showed that the FPA protein played a wide role in seed development involving multiple developmental processes. In addition, 1064 transposable element genes were detected in seed from RNA sequencing. Among these genes, 43 transposable element genes were significantly differentially expressed and upregulated in spz2 zou-4 (Supplementary Table 2).
Discussion
It is believed that intronic T-DNA insertions often produce loss of function alleles because of improper splicing and/or premature mRNA 3′-end formation (Wang 2008). Until now, little was known about how genes control the effects of intronic T-DNA insertions. From the study of the spz2 mutation, we first inferred that the FPA may be an important regulatory factor. In the zou-4 mutant, the chimeric transcripts were prematurely terminated at proximal polyadenylation sites, whereas in the spz2 zou-4 mutant, the premature termination was reduced, resulting in read-through of the T-DNA and eventually removal of the T-DNA sequences together with the surrounding intron during splicing. This indicated that FPA can promote the use of the proximal polyadenylation site to generate truncated transcripts in a T-DNA insertion mutation. This role of FPA in regulating an intronic T-DNA mutation is entirely consistent with its function as a trans-acting regulator of poly(A) site choice and alternative polyadenylation (APA; Hornyik et al. 2010; Duc et al. 2013).
Global analyses have revealed that about 70 % of eukaryotic genes produce multiple mRNA isoforms with distinct 3′-ends through the process of alternative polyadenylation (APA; Di Giammartino et al. 2011; Shi 2012; Elkon et al. 2013; Tian and Manley 2013). APA is dynamically regulated in development and in response to environmental stimuli (Sandberg et al. 2008; Shepard et al. 2011; Graber et al. 2013), and deregulation of APA has been associated with a number of human diseases (Mayr and Bartel 2009; Jenal et al. 2012; Masamha et al. 2014). The mechanisms governing APA have only recently begun to be understood. Variations in the levels of the core polyadenylation factors and splicing factors or abundance of a broad range of transacting RNA-binding proteins have been shown to modulate PAS selection (Elkon et al. 2013; Shi and Manley 2015). In addition, emerging evidence suggests that APA is also influenced by chromatin organization and epigenetic modifications at the DNA level (Spies et al. 2009). Here, a T-DNA insertion in the zou-4 mutant produces three proximal polyadenylation sites, including one in the native ZOU first intron and two in the T-DNA sequence. However, the cryptic polyadenylation site in the ZOU first intron is not normally recognized in the wild type. This suggested that the T-DNA insertion can change the polyadenylation property and produce novel polyadenylation sites recognized by FPA.
The suppression of intronic T-DNA mutation by fpa mutations was only observed in the zou-4 background and not in the zou-3 background even though the zou-3 mutant is also an intronic T-DNA insertion mutant with the T-DNA inserted in the first intron very near the T-DNA insertion site of zou-4 (Supplementary Fig. S4). The zou-4 mutant was obtained from the GABI-KAT collection generated using the T-DNA vector pAC161, whereas the zou-3 mutant was from the University of Wisconsin collection of T-DNA insertion lines mutated using the T-DNA vector pDs-Lox. The different T-DNA insertion vectors present at zou-4 and zou-3 seem likely to be the major reason for the different response to fpa mutations. The T-DNA in zou-3 contained a 35S polyadenylation motif behind the LB insertion site, which is the candidate element contributing to obtaining different phenotypes between zou-3 and zou-4, for example; since this may be a strong termination site, it may be less dependent on FPA activity for its selection.
Methods
Plant materials and growth conditions
The zou-4 (Col-0 ecotype) allele was obtained from the Gabi-Kat collection of T-DNA inserts (Rosso et al. 2003) and corresponded to GABI_584D09. The zou-2 allele (Ws-0 ecotype) arose in the FLAG collection of T-DNA insertions (Samson et al. 2002) and corresponded to FLAG 400A08. The zou-3 allele (Col-0 ecotype) was obtained from the University of Wisconsin collection of T-DNA insertion lines (Sussman et al. 2000) and corresponded to WiscDsLox465F5. The spz mutants in the zou-4 background were backcrossed six times to zou-4 before analysis.
Unless noted otherwise, the seeds were sterilized and germinated on sterile tissue culture medium comprising 1/2 MS salts (Sigma), 0.3 % sucrose and 0.8 % agar, stored at 4 °C for 3 days. Seedlings were transferred to the soil about 10 days after germination and grown under long-day conditions (16 h light/8 h dark) at 22 °C with a light intensity of 120 μmol photons m−2 s−1.
Map-based cloning and allelic analysis
The spz2 zou-4 mutant from the Col-0 ecotype was crossed with zou-2 in the Ws-0 ecotype for positional cloning. After selfing of F1 plants, the homozygote mutants with a full seed phenotype from the F2 population were selected for mapping. The spz2 mutation was preliminarily mapped with Simple Sequence Length Polymorphism (SSLP) molecular markers (TAIR; http://www.Arabidopsis.org). For spz2 fine mapping, plants were genotyped with SSLP and derived Cleaved Amplified Polymorphic Sequence (dCAPS) markers, designed based on the Col-0 and Ws-0 ectype Arabidopsis genomic sequence (Gan et al. 2011). The interval was narrowed down to 75 kb on chromosome 2. Finally, the complete 75-kb genomic region was sequenced. The primers used in fine mapping and gene cloning are listed in Supplementary Table 3.
In the allelic analysis, the spz1-23 mutants were crossed with spz2 zou-4. Two allelic mutants (spz10 and spz23) were identified according to the lack of complementation, i.e., the F1 seeds had a full seed phenotype. The lesions responsible for these allelic mutants were determined by sequencing. Primers used to examine the mutation sites are listed in Supplementary Table 4.
Vector construction
For complementation, spz2 was transformed with binary vector ZYH168. The 7.7-kb genomic SPZ2 fragment starting from 1582 bp upstream of the ATG of SPZ2 to 1562 bp downstream from the stop codon was PCR amplified from Col-0 using Phusion High-Fidelity PCR Master Mix (NEB) with primers OL1780 and OL1784 (Supplementary Table 4). The PCR fragments were given a 3′-A overhang by treatment with EasyTaq and dATP (TransGenBiotech) and then cloned into the pMD18-T vector (Takara) to generate the construct ZYH165 containing the genomic fragment in the desired orientation. The ZYH165 construct was digested with XbaI and cloned into the binary vector pCAMBIA3301 (Cambia), generating the complementation construct ZYH168.
For the GUS reporter construct, the SPZ2 promoter fragment extending from 2373 bp upstream of the ATG to 157 bp downstream of the ATG was amplified by PrimeSTAR HS DNA Polymerase (Takara) using the primers OL1857 and OL1858 (Table S4). The PCR fragments were given a 3′-A overhang by treatment with EasyTaq (TransGenBiotech) and directly cloned into the binary vector pCXGUS-P (Chen et al. 2009) to generate the GUS reporter construct ZYH155.
Histochemical GUS analysis
Histochemical staining for GUS reporter gene activity was performed as described previously (Stangeland and Salehian 2002; Thines et al. 2007). Seedlings at 3, 14 and 21 days after germination and seeds at 2, 5, 7 and 9 days after pollination were immersed in 90 % acetone (chilled) and kept on ice until all samples were collected. Samples were incubated for 20 min at room temperature and then washed with a solution of 0.05 M sodium phosphate buffer, pH 7.2, 0.2 % Triton X-100, 2 mM potassium ferrocyanide and 2 mM potassium ferricyanide. Washing solution was removed and staining solution (washing solution plus 2 mM X-Gluc) was added, and the samples were incubated overnight at 37 °C. After incubation, the staining solution was removed, and the samples were subject to a series of ethanol washes: 20, 35, and 50 % at room temperature for 30 min each. The seeds were then cleared in modified Hoyer’s solution (chloral hydrate:water:glycerol in proportions 8 g:2 ml:1 ml) and visualized under a Nikon microscope using DIC optics.
RNA extraction and analysis of gene expression
Total RNA from Arabidopsis developing seeds was extracted according to Oñate-Sánchez and Vicente-Carbajosa (2008). The extracted RNA was subjected to DNase I treatment (Takara). First-strand cDNA was prepared from 0.5 μg total RNA using the AMV First Strand cDNA Synthesis Kit (NEB) according to the manufacturer’s instructions. PCR reactions were performed in triplicate. The products were quantified using a Bio-Rad DNA Engine Opticon 2 real-time PCR machine, and the associated software was used to assay SYBR green fluorescence. PCR reactions were performed with FastStart Universal SYBR Green Master (Roche). The primers used are listed in Supplementary Table 4.
The 3′-race assay
The 3′-RACE assay was performed using the SMARTer RACE cDNA Amplification Kit (Clontech). Briefly, total RNA was extracted from Col-0, zou-4 and spz2 zou-4 mutant seeds, and the extracted RNA was subjected to DNase I treatment (Takara). First-strand cDNA was reverse transcribed from 2 μg of total RNA with the 3′-RACE coding sequence (CDS) Primer A (provided in the kit). The PCR was performed with gene-specific primers (OL3809) and the Universal Primer Mix (provided by the kit). The PCR products were detected by southern blot with probes in ZOU exon 1 (amplified by primers OL3789 and OL3792) (Supplementary Table 4) and subsequently cloned into the pRACE vector by In-fusion cloning (Clontech) and sequenced.
RNA sequencing and transcriptome analysis
The seeds from Col-0, zou-4 and spz2 zou-4 at the heart stage about 5 days after pollination were picked out from siliques. After extracting the total RNA from the seeds, mRNA was enriched by using oligo(dT) magnetic beads (Ambion). The mRNA was fragmented into short fragments (about 200 bp), and then first-strand cDNA was synthesized with random hexamer primers using the mRNA fragments as templates, and adaptors were then ligated to the fragments. The required fragments were purified by agrose gel electrophoresis and enriched by PCR amplification. The library products were sequenced via Illumina HiSeq™ 2000. The genes with significant expression differentiation were filtered according to FDR ≤ 0.001 and |log2Ratio| ≥ 1, which measured the significance of the difference between two samples’ PRKM value. GO analysis used WEGO according to Ye et al. (2006).
References
Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R et al (2003) Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301:653–657
Bäurle I, Smith L, Baulcombe DC, Dean C (2007) Widespread role for the flowering-time regulators FCA and FPA in RNA-mediated chromatin silencing. Science 318:109–112
Chen S, Songkumarn P, Liu J, Wang GL (2009) A versatile zero background T-vector system for gene cloning and functional genomics. Plant Physiol 150:1111–1121
Chinnusamy V, Ohta M, Kanrar S, Lee BH, Hong X, Agarwal M, Zhu JK (2003) ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev 17:1043–1054
Denay G, Creff A, Moussu S, Wagnon P, Thévenin J, Gérentes MF, Chambrier P, Dubreucq B, Ingram G (2014) Endosperm breakdown in Arabidopsis requires heterodimers of the basic helix-loop-helix proteins ZHOUPI and INDUCER OF CBP EXPRESSION 1. Development 141:1222–1227
Di Giammartino DC, Nishida K, Manley JL (2011) Mechanisms and consequences of alternative polyadenylation. Mol Cell 43:853–866
Duc C, Sherstnev A, Cole C, Barton GJ, Simpson GG (2013) Transcription termination and chimeric RNA formation controlled by Arabidopsis thaliana FPA. PLoS Genet 9:e1003867
Elkon R, Ugalde AP, Agami R (2013) Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet 14:496–506
Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT et al (2011) Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477:419–423
Gao Y, Zhao Y (2013) Epigenetic suppression of T-DNA insertion mutants in Arabidopsis. Mol Plant 6:539–545
Graber JH, Nazeer FI, Yeh PC, Kuehner JN, Borikar S, Hoskinson D, Moore CL (2013) DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast. Genome Res 23:1690–1703
Hornyik C, Terzi LC, Simpson GG (2010) The spen family protein FPA controls alternative cleavage and polyadenylation of RNA. Dev Cell 18:203–213
Ingram GC (2010) Family life at close quarters: communication and constraint in angiosperm seed development. Protoplasma 247:195–214
Jenal M, Elkon R, Loayza-Puch F, van Haaften G, Kuhn U, Menzies FM, Oude Vrielink JA, Bos AJ, Drost J, Rooijers K et al (2012) The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 149:538–553
Jia X, Chanda B, Zhao M, Brunner AM, Beers EP (2015) Instability of the Arabidopsis mutant csn5a-2 caused by epigenetic modification of intronic T-DNA. Plant Sci 238:53–63
Kanaoka MM, Pillitteri LJ, Fujii H, Yoshida Y, Bogenschutz NL, Takabayashi J, Zhu JK, Torii KU (2008) SCREAM/ICE1 and SCREAM2 specify three cell-state transitional steps leading to Arabidopsis stomatal differentiation. Plant Cell 20:1775–1785
Kondou Y, Nakazawa M, Kawashima M, Ichikawa T, Yoshizumi T, Suzuki K, Ishikawa A, Koshi T, Matsui R, Muto S, Matsui M (2008) RETARDED GROWTH OF EMBRYO1, a new basic helix-loop-helix protein, expresses in endosperm to control embryo growth. Plant Physiol 147:1924–1935
Krysan PJ, Young JC, Sussman MR (1999) T-DNA as an insertional mutagen in Arabidopsis. Plant Cell 11:2283–2290
Lee BH, Henderson DA, Zhu JK (2005) The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 17:3155–3175
Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ (2014) CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510:412–416
Mayr C, Bartel DP (2009) Wide-spread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138:673–684
Miura K, Jin JB, Lee J, Yoo CY, Stirm V, Miura T, Ashworth EN, Bressan RA, Yun DJ, Hasegawa PM (2007) SIZ1-mediated sumoylation of ICE1 controls CBF3/DREB1A expression and freezing tolerance in Arabidopsis. Plant Cell 19:1403–1414
Olsen OA (2004) Nuclear endosperm development in Cereals and Arabidopsis thaliana. Plant Cell 16:S214–S227
Oñate-Sánchez L, Vicente-Carbajosa J (2008) DNA-free RNA isolation protocols for Arabidopsis thaliana, including seeds and siliques. BMC Res Notes 1:93
Rosso MG, Li Y, Strizhov N, Reiss B, Dekker K, Weisshaar B (2003) An Arabidopsis thaliana T-DNA mutagenized population (GABI-Kat) for flanking sequence tag-based reverse genetics. Plant Mol Biol 53:247–259
Samson F, Brunaud V, Balzergue S, Dubreucq B, Lepiniec L, Pelletier G, Caboche M, Lecharny A (2002) FLAGdb/FST: a database of mapped flanking insertion sites (FSTs) of Arabidopsis thaliana T-DNA transformants. Nucleic Acids Res 30:94–97
Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB (2008) Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320:1643–1647
Sandhu KS, Koirala PS, Neff MM (2013) The ben1-1 brassinosteroid-catabolism mutation is unstable due to epigenetic modifications of the intronic T-DNA insertion. G3 3:1587–1595
Schomburg FM, Patton DA, Meinke DW, Amasino RM (2001) FPA, a gene involved in floral induction in Arabidopsis, encodes a protein containing RNA-recognition motifs. Plant Cell 13:1427–1436
Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, Shi Y (2011) Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17:761–772
Shi Y (2012) Alternative polyadenylation: new insights from global analyses. RNA 18:2105–2117
Shi Y, Manley JL (2015) The end of the message: multiple protein–RNA interactions define the mRNA polyadenylation site. Genes Dev 29:889–897
Spies N, Nielsen CB, Padgett RA, Burge CB (2009) Biased chromatin signatures around polyadenylation sites and exons. Mol Cell 36:245–254
Stangeland B, Salehian Z (2002) An improved clearing method for GUS assay in Arabidopsis endosperm and seeds. Plant Mol Bio Rep 20:107–114
Stroud H, Greenberg MVC, Feng S, Bernatavichute YV, Jacobsen SE (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152:352–364
Sussman MR, Amasino RM, Young JC, Krysan PJ, Austin-Phillips S (2000) The Arabidopsis knockout facility at the University of Wisconsin-Madison. Plant Physiol 124:1465–1467
Tanaka H, Onouchi H, Kondo M, Hara-Nishimura I, Nishimura M, Machida C, Machida Y (2001) A subtilisin-like serine protease is required for epidermal surface formation in Arabidopsis embryos and juvenile plants. Development 128:4681–4689
Thines B, Katsir L, Melotto M, Niu Y, Mandaokar A, Liu G, Nomura K, He SY, Howe GA, Browse J (2007) JAZ repressor proteins are targets of the SCFCOI1 complex during jasmonate signaling. Nature 448:661–665
Tian B, Manley JL (2013) Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci 38:312–320
Ulker B, Peiter E, Dixon DP, Moffat C, Capper R, Bouché N, Edwards R, Sanders D, Knight H, Knight MR (2008) Getting the most out of publicly available T-DNA insertion lines. Plant J 56:665–677
Wang YH (2008) How effective is T-DNA insertional mutagenesis in Arabidopsis. J Biochem Technol 1:11–20
Xiang Y, Nakabayashi K, Ding J, He F, Bentsink L, Soppe WJJ (2014) Reduced Dormancy5 encodes a protein phosphatase 2C that is required for seed dormancy in Arabidopsis. Plant Cell 26:4362–4375
Xing Q, Creff A, Waters A, Tanaka H, Goodrich J, Ingram GC (2013) ZHOUPI controls embryonic cuticle formation via a signalling pathway involving the subtilisin protease ABNORMAL LEAFSHAPE1 and the receptor kinases GASSHO1 and GASSHO2. Development 140:770–779
Xue W, Ruprecht C, Street N, Hematy K, Chang C, Frommer WB, Persson S, Niittyla T (2012) Paramutation-like interaction of T-DNA loci in Arabidopsis. PLoS ONE 7:e51651
Yang S, Johnston N, Talideh E, Mitchell S, Jeffree C, Goodrich J, Ingram G (2008) The endosperm-specific ZHOUPI gene of Arabidopsis thaliana regulates endosperm breakdown and embryonic epidermal development. Development 135:3501–3509
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:293–297
Acknowledgments
This work was supported by the National Nature Science Foundation of China (grant nos. 31470286, 30970278 and 31271743) and the China National Transgenic Major Program (grant no. 2014ZX0800943B) and the Natural Science Foundation of Shandong Province of China (grant no. ZR2011CM020) and was also supported by the One Hundred Person Project of the Chinese Academy of Sciences.
Author contributions
S.X.Y., X.Z.F. and J.G. conceived the project and designed this work. Y.H.Z. and X.L. constructed the mutation library and performed map-based cloning. Y.H.Z., X.L. and C.X.W. performed transgenic, cell biological and other functional analyses. H.C.W. performed RNA sequencing analysis. S.X.Y., X.Z.F. and J.G. wrote this paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Yaohua Zhang and Xin Li contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Y., Li, X., Goodrich, J. et al. Reduced function of the RNA-binding protein FPA rescues a T-DNA insertion mutant in the Arabidopsis ZHOUPI gene by promoting transcriptional read-through. Plant Mol Biol 91, 549–561 (2016). https://doi.org/10.1007/s11103-016-0487-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-016-0487-2