
Abstract
Purpose
This study investigates the cellular uptake and trafficking of liposomes in Caco-2 cells, using vesicles with distinct average diameters ranging from 40.6 nm to 276.6 nm. Liposomes were prepared by microfluidic hydrodynamic flow focusing, producing nearly-monodisperse populations and enabling size-dependent uptake to be effectively evaluated.
Methods
Populations of PEG-conjugated liposomes of various distinct sizes were prepared in a disposable microfluidic device using a simple continuous-flow microfluidic technique. Liposome cellular uptake was investigated using flow cytometry and confocal microscopy.
Results
Liposome uptake by Caco-2 cells was observed to be strongly size-dependent for liposomes with mean diameters ranging from 40.6 nm to 276.6 nm. When testing these liposomes against endocytosis inhibitors, cellular uptake of the largest (97.8 nm and 162.1 nm in diameter) liposomes were predominantly subjected to clathrin-dependent uptake mechanisms, the medium-sized (72.3 nm in diameter) liposomes seemed to be influenced by all investigated pathways and the smallest liposomes (40.6 nm in diameter) primarily followed a dynamin-dependent pathway. In addition, the 40.6 nm, 72.3 nm, and 162.1 nm diameter liposomes showed slightly decreased accumulation within endosomes after 1 h compared to liposomes which were 97.8 nm in diameter. Conversely, liposome co-localization with lysosomes was consistent for liposomes ranging from 40.6 nm to 97.8 nm in diameter.
Conclusions
The continuous-flow synthesis of nearly-monodisperse populations of liposomes of distinct size via a microfluidic hydrodynamic flow focusing technique enabled unique in vitro studies in which specific effects of particle size on cellular uptake were elucidated. The results of this study highlight the significant influence of liposome size on cellular uptake mechanisms and may be further exploited for increasing specificity, improving efficacy, and reducing toxicity of liposomal drug delivery systems.
Similar content being viewed by others
Introduction
Liposomes have received a great deal of attention as drug delivery vehicles owing to their ability to transport a range of therapeutic agents. Such advances in liposome uses have been highlighted and discussed within review papers (1–4). Currently, the most widespread form of liposomal drug delivery is Doxil, which is the first FDA approved liposome based drug used in anti-cancer therapy (5,6). While the utility of liposomes as delivery vehicles for therapeutics and vaccines has been widely recognized, there remain many hurdles for effective delivery to targeted tissues and cells (7). In particular, current liposome preparation techniques rely on bulk-scale synthesis and typically result in a limited range of size and uniformity (8). Due to the inevitable size variability within a single population of liposomes, determination of an efficient size-based targeting mechanism remains a challenge. Liposome size and variability in size are important factors that can influence its drug dosage, targeting and clearance (9,10). Initial cellular recognition of nanoparticles used in drug delivery depends on particle surface chemistry, size, and morphology (11–14). Size has also been shown to be an important factor for efficient cellular uptake and extended circulation time (9,10), hence the reduction in size variability has the potential to improve performance in liposomal drug delivery systems.
Established bulk-scale processes offer limited ability to produce monodisperse liposomes within the size range deemed acceptable for nanoparticle drug carriers (50 nm to 200 nm in diameter) (15). Liposome populations produced from bulk-scale techniques encompass a great deal of variability in average diameter and homogeneity(16–20), even after multiple post-processing steps to reduce the range of size distribution. In contrast, a demonstrated microfluidic-based technique for liposome synthesis presents a new avenue for producing populations of liposomes with tunable size and exceptionally low polydispersity compared to traditional methods (21–23). The microfluidic method is an implementation of the alcohol-injection technique for liposome production within a microfluidic network, exploiting hydrodynamic focusing for controlled diffusive mixing of chemical species at the nanoscale to enable precise self-assembly of lipids into liposomes whose sizes may be dynamically adjusted by simply modifying the flow conditions within the device. Unlike traditional methods, whose bulk-scale synthesis processes rely on turbulent flows to induce liposome self-assembly in an unpredictable manner and are thus followed by requisite additional processing steps for size homogenization, the microfluidic technique exploits exquisitely controlled diffusive transport and laminar convective flows within a microfluidic channel network to enable one-step, continuous-flow liposome production without post-processing steps for further size regularization. This technique is a significant advancement in the preparation of liposomes, enabling formation of liposomes ranging from 30 nm to several hundred nanometers with minimal size variability within a given population (21).
Here, we have investigated how endocytosis behavior in Caco-2 cells is affected by particle size by using liposome synthesized through the microfluidic technique, thus enabling inspection of each phenomenon with much finer resolution than studies involving traditional preparation methods. Cellular uptake mechanisms were investigated through the use of various endocytosis inhibitors, in addition to intracellular trafficking of varied liposome vesicle size was investigated over time using confocal microscopy, distinguishing the internalization pathways and uptake mechanisms used by the Caco-2 cells upon exposure to liposomes of various sizes.
Materials and Methods
Microfluidic Preparation of Liposomes
Device Fabrication
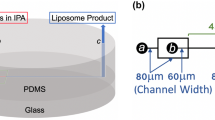
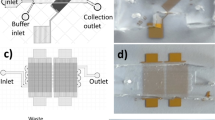
Microfluidic devices were fabricated using soft lithography techniques. Microchannel designs were created using AutoCAD software (Autodesk, San Rafael, CA) and printed onto a photomask (Fineline Imaging, Colorado Springs, CO). To create features for imprinting microchannels, SU-8 negative photoresist (MicroChem Corp., Newton, MA) was spin-coated onto a 4-inch silicon wafer (University Wafer, South Boston, MA), exposed to ultraviolet light through the photomask on an automated EVG 620 mask aligner (EV Group, Germany), and developed to establish a master mold with raised features which may be used repeatedly to create microchannels in poly(dimethylsiloxane) (PDMS) elastomer. The SU-8 mold was placed in a plastic petri dish and a 10:1 (w:w) mixture of pre-polymer PDMS elastomer and curing agent (Sylgard 184, Dow Corning Corp. Midland, MI) was poured over the mold. Vacuum was applied to the petri dish to remove air bubbles before being placed in a convection oven at 80°C for 4 h to ensure complete curing of the PDMS. The PDMS was carefully removed from the SU-8 mold and sectioned into devices by a scalpel. Inlet and outlet holes were made using a biopsy punch (Harris Uni-Core, Ted Pella, Inc., Redding, CA). The bonding surfaces of the PDMS and a glass slide were cleaned using isopropanol and DI water then exposed to oxygen plasma in a March Jupiter III Reactive Ion Etcher (Nordson Corp., Concord, CA). The PDMS and glass pieces were mated and placed in a convection oven at 80°C for 2 h for bonding to occur. Microfluidic device channels were 15 μm wide and 150 μm high.
Lipid Mixture and Hydration Buffer Preparation
Dipalmitoylphosphatidylcholine (DPPC), cholesterol, and dipalmitoylphosphatidylethanolamine-poly(ethylene glycol) 2000 (PEG2000-PE) (all from Avanti Polar Lipids Inc., Alabaster, AL) were mixed in chloroform (Mallinckrodt Baker Inc., Phillipsburg, NJ) at a molar ratio of 5:4:1, respectively. The lipid mixture was prepared in glass scintillation vials then placed in a vacuum desiccator for at least 24 h to ensure complete solvent removal. The dried lipid mixtures were then redissolved in anhydrous ethanol (Sigma Aldrich) with 1 wt% of a lipophilic membrane dye, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI-C18) (Life Technologies, Carlsbad, CA), Carlsbad, CA)) for a total lipid concentration of 15 mM. A 10 mM phosphate buffered saline (PBS) (Sigma Aldrich) solution at pH 7.4 was used as a hydration buffer. All fluids (solvent and buffer) were passed through 0.22 μm filters (Millipore Corp., New Bedford, MA) before being introduced to the microfluidic device.
Microfluidic Liposome Synthesis
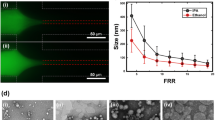
The PDMS-glass microfluidic devices were used to form PEGylated liposomes by including PEG-conjugated lipids during liposome synthesis with phosphate buffered saline (PBS) as a hydration buffer using the method demonstrated previous (24). Briefly, PBS was injected into two side channels intersecting with a center channel containing the ethanol/lipid mixture. The flow rate ratio (FRR), defined as the ratio of volumetric flow rate of aqueous buffer to that of solvent, was varied from 3-15 to produce liposomes of various sizes. The linear flow velocity of the combined fluid streams for all FRRs was kept constant at 0.20 m/s. The hydrodynamic focusing region within the microfluidic device was monitored with a TE-2000 S epifluorescence inverted microscope (Nikon, Melville, NY) during liposome formation to ensure the presence of consistent and symmetric flow conditions.
Liposome Characterization via Asymmetric Flow Field-Flow Fractionation with MALLS and QELS
High-resolution size-based separation of the liposome populations was carried out with Asymmetric Flow Field-Flow Fractionation (AF4). PBS buffer was used for the AF4 separations, which were combined in-line with multi-angle laser light scattering (MALLS) and quasi-elastic light scattering (QELS) for liposome detection and characterization (DAWN HELEOS and QELS, Wyatt Technology, Santa Barbara, CA). A vendor-supplied spacer (250 μm thickness) was used to define the flow channel thickness with a 10 kDa molecular weight cut off (MWCO) regenerated cellulose membrane for the cross-flow partition (Millipore, Bedford, MA). The flow was controlled with Eclipse 2 software (Wyatt Technology, Santa Barbara, CA). A sample volume of 10 μL of each liposome population was injected at a flow rate of 0.1 μL/min while focusing at 1.5 mL/min for 5 min. The injection step was succeeded by a second focusing step of 1.5 mL/min for 5 min. The crossflow was linearly decreased from 0.2 to 0 mL/min over an 18 min while eluting the separated particles at 1 mL/min. The radii of the separated particles were determined using the MALLS and QELS detectors with data processing using ASTRA software (Wyatt Technology, Santa Barbara, CA). Static light scattering intensity (λ = 690 nm) was measured at 15 angles simultaneously. Data was collected at 1 Hz by MALLS and 0.2 Hz by QELS. The autocorrelation function of the QELS was fitted to a single-mode exponential decay model to calculate the hydrodynamic radii of the liposomes. A coated-sphere light scattering model (i.e. a spherical structure with two radial regions of differing refractive index) was used to calculate the geometric radii of the fractionated liposome samples.
Cellular Uptake Studies
Caco-2 Cell Culture
Caco-2 cells were cultured at 37°C in an atmosphere of 95% relative humidity and 5% CO2 (vol%). Cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acids, 10,000 units/mL penicillin, 10,000 μg/mL streptomycin and 25 μg/mL amphotericin B. Media was changed every second day and cells were passaged at approximately 90% confluence using a 0.25% trypsin/ethylendiamine tetraacetic acid (EDTA) solution. Caco-2 cells were obtained from American Type Culture Collection (ATCC)(Rockville, MD)
Cytotoxicity of Endocytosis Inhibitors
Cytotoxicity of endocytosis inhibitors was assessed in Caco-2 cells to ensure the cell viability over short-term exposure to these chemicals during uptake experiments. Endocytosis inhibitors were prepared at a range of concentrations known to reduce the different pathways (purchased from Sigma Aldrich, St. Louis, MO). Inhibitors used for their respective pathways were as follows: monodansyl cadaverine (MDC) (150 μM to 600 μM) for clathrin mediated endocytosis; filipin (FIL) (2 μM to 8 μM) for reduction of caveolin-mediated endocytosis; dynasore (DYN) (25 μM to 100 μM) for dynamin-dependent endocytosis; and wortmannin (WORT) (50 nM to 200 nM) for macropinocytosis (The range of all the inhibitors was determined through literature (25–27) Cytotoxicity of the inhibitors was assessed by the water-soluble tetrazolium salt (WST-1) assay (Roche Applied Science, Indianapolis, IN). Caco-2 cells were seeded at 50,000 cells per well in a 96-well plate (Corning, NY). Cells were incubated at 37°C, 95% relative humidity and 5% CO2 for 48 h. Cells were washed with warm Hank’s balanced salt solution (HBSS) buffer and incubated for 2 h with 100 μL solutions containing a varied concentration of endocytosis inhibitors. The solutions were removed after 2 h and cells were washed twice with HBSS buffer. WST-1 assay reagent solution was added to each well (10 μL of WST-1 in 100 μL of HBSS for each well) and incubated for 4 h at 37°C. Following the incubation time, the plate was mixed well and then measured for absorbance on a plate reader. Absorbance at 460 nm and background at 600 nm were measured using a SpectraMax plate reader (Molecular Devices, Sunnyvale, CA). Cells incubated in HBSS were used as the negative control for 100% viability and cells incubated in Triton-X were used as the positive control. Cell viability ≥80% was classified as concentrations acceptable for uptake studies. As represented in Table II, MDC at 300 μM, WORT at 100 nM, FIL 4 μM and DYN at 50 μM were used for the cellular uptake studies.
Cellular Uptake
Cellular uptake of different sized liposomes was determined in the presence and absence of endocytosis inhibitors. Inhibitors used were at concentrations that showed a minimum of 85% of viability during the 2 h period of the assay incubation period. Cells were seeded at 150,000 cell/well in a 12-well plate (Corning, NY) and grown for 3 days - 4 days at 37°C, 95% relative humidity and 5% CO2. Culture medium was then removed and cells were washed twice with warm HBSS. The required amount of HBSS and liposome solution was added to each well such that the total volume was 400 μL. Cells were incubated with the liposome solution for 15 min and 1 h at 37°C. The liposome mixture was then removed and cells were washed twice with a 1 mL solution of ice-cold Dulbecco’s phosphate buffered saline (DPBS). Cells were then incubated with trypsin for 5 min after which cell culture medium was added to halt this detachment process. Cells were removed from plates, transferred to microcentrifuge tubes, and centrifuged for 4 min at 1500 rpm. After removing the supernatant, cells were washed in DPBS and finally fixed in 1% (wt%) paraformaldehyde (in DPBS) solution. Flow cytometry was used to measure the cellular fluorescence using the BD LSR flow cytometer (Becton Dickenson, Franklin Lakes, NJ) with filters, Ex - 552 nm; Em - 577/10 nm. A 200 μL solution of 0.4% (wt%) Trypan Blue was added to each sample and 10,000 events to 20,000 events were taken per sample for 4 repeat experimental samples. Percentage uptake was determined for different cell populations by the shift in mean fluorescence (region of interest was determined using FlowJo software) in the presence of endocytosis inhibitors compared to the controls in HBSS. Uptake analysis for the influence of liposome size on cell populations was determined by comparing the shift in means between each liposome size sample. The data for fluorescence intensity events for all liposome sizes recorded by flow cytometry was normalized to the total uptake of liposomes at 37°C in the absence of any inhibitor (this gives the 100% value on the graph) and to the difference between the uptake in presence of inhibitors by its uptake value at 4°C (for each liposome size the 4°C uptake was different since this represents the passive uptake and hence highlights the region of interest). Hence the values presented in Fig. 4 represent the percentage of liposomes not affected by the inhibitor.
Intracellular Co-localization
Intracellular trafficking of liposomes of different sizes within Caco-2 cells was also investigated in addition to their uptake mechanism. Caco-2 cells were seeded at 5000 cell/cm2 on 8-chamber slides. Slides were used after 3 days to 5 days of incubation at 37°C, 95% relative humidity and 5% CO2. Cells were washed with warm HBSS and then incubated with liposomes in HBSS for 15 min to 1 h. After which cells were washed with ice-cold DPBS, and fixed with 4% (wt%) paraformaldehyde, 4% (wt%) sucrose in DPBS for 15 min. Cells were washed with permeabilization buffer (PBS containing 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2 · 6H2O, 20 mM HEPES, 0.5% Triton X 100 (vol%), pH 7.2), which was added to the samples and kept at 4°C for 5 min. The permeabilization buffer washed out twice with DPBS and later incubated at 37°C with blocking buffer (3% bovine serum albumin (BSA) (wt/vol) in DPBS) for 5 min. Cells were washed with DPBS and primary antibodies for early endosomes (rabbit polyclonal early endosome antigen -1 (EEA-1; Molecular Probes) and lysosome-associated membrane protein 1 (rabbit polyclonal lysosome antigen – 1 (LAMP-1); Molecular Probes) (both solutions prepared at 1:500 vol.ratio in 3% BSA solution) were added to separate cell sample chambers and left for 1 h in the incubator at 37°C. The primary antibody was then removed and the cells were washed 3 times with blocking buffer and then the secondary antibody Alexa flour-488 goat anti-rabbit IgG (Molecular probes, Carlsbad, CA) was added at 1:1000 in the blocking agent solution for 1 h. The cells were then washed with 0.5 vol (vol%) Tween 20 in DPBS and finally three times with DPBS. Cells were then incubated with 300 nM 4’,6-diamidino-2-phenylindole (DAPI) for 10 min to stain the nuclei. The cells were then washed twice with DPBS and the chambers were removed. The slides were mounted and covered with glass coverslips. They were allowed to dry for a few hours before they were sealed with nail varnish and stored at 4°C. Images were acquired using a Nikon A1 Inverted confocal laser-scanning microscope (Nikon Instruments, Melville, NY). Co-localization between liposomes with early endosomes and lysosomes was quantified using the Elements software supplied with the Nikon microscope. The extent of co-localization between the overlapping channels was determined using the Mander’s overlap coefficient (Mx(oc)). The extent of co-localization between the red and green channels (Mx(oc)) was calculated using the following equation:
where xi, coloc is the value of voxels of the overlapped red with green components, and xi is the value of the red component. Mx(oc) is reported for each treatment as an average of the 6 images where the threshold was adjusted to 5% for all images, in order to compensate for any background noise. This method of calculating the co-localization was adopted from a previously published journal article (26), however here we used Nikon Elements for acquisition and the Volocity 3D imaging software (Improvision, Lexington, MA) to determine our co-localization values.
Results
Microfluidic Liposome Synthesis
Liposome solutions prepared by microfluidic flow focusing were characterized using MALLS and QELS in-line with AF4 to determine liposome diameter, size distribution, and final number concentration of liposomes in solution. The mean diameters of liposome populations used for cellular uptake experiments ranged from 40.6 nm to 276.6 nm (Fig. 1, Table I). The relationship between FRR and average liposome diameter of populations made within the PDMS-glass device followed trends of previously demonstrated studies (22,28), showing a decrease in liposome size corresponding to an increase in FRR. The populations of liposomes created for this study exhibited low levels of polydispersity with very distinct diameters.

Diameter range determined by MALLS and QELS in line with AF4. Data show size and size distribution of liposomes (in nm) prepared by microfluidics. Mean diameter for liposomes is presented with ± distribution of diameters (nm) for each sample. Liposome populations represented here are referenced by modal diameter throughout the text.
Cellular Uptake Studies
Cytotoxicity of Liposome Populations
Cytotoxicity tests were performed to ensure that toxicity of liposomes would have no influence on cellular uptake. The WST-1 reagent assay was used to examine the viability of Caco-2 cells in presence of the differently sized liposomes at varied concentrations. Caco-2 cells, seeded at 5 × 104 cells/well, were incubated for 2 days at 37°C, 95% relative humidity and 5% CO2. Cells were incubated with the test liposome solution and assessed by the WST-1 assay at different concentrations. 90% to 95% viability was chosen as the minimum allowable concentration for use in uptake studies.
Size Dependent Uptake of Liposomes in Caco-2 Cells
Caco-2 cells were incubated with liposomes, with the number of liposomes introduced from each sample held constant. The values for total cellular uptake were determined by flow cytometry with the signal normalized to liposome surface area (DiI dye is encapsulated within the liposomal bilayer). The resulting fluorescence data from flow cytometry indicated that cellular uptake was dependent on liposome size (Fig. 2). Liposome uptake was measured for two different incubation time points, 15 min and 1 h, both at 37°C. The total cellular uptake for the smallest liposomes (40.6 nm) was significantly higher, almost a 12-fold increase, than the largest liposomes (162.1 nm and 276.6 nm). Cellular uptake of the medium-sized liposomes (86.2 nm and 97.8 nm) demonstrated an intermediate level of uptake compared to the liposomes on opposite ends of this size spectrum.

Size dependent uptake of liposomes. Flow cytometry results for Caco-2 cells incubated for 15 min and 1 h at 37°C with liposomes ranging from 40.6 nm to 276.6 nm in mean diameter containing DiI-C18 lipophilic dye. Fluorescence data was normalized to liposome diameter, revealing the highest liposome uptake for 40.6 nm. Each cell population was incubated with a consistent particle concentration (2 × 1010 liposomes/mL). *** indicates a significant difference (p < 0.001) by One-way ANOVA test when compared with control sample without liposomes.
Endocytosis Inhibitors and Their Influence on Liposome Uptake
Four endocytosis inhibitors were used to examine the pathways of cellular uptake as a function of liposome size. Before initiating the uptake and transport studies, Caco-2 cell viability during 2 h assay time was confirmed in the presence of the endocytosis inhibitors and the concentrations were determined based on those found in literature (25–27). The inhibitor concentrations that showed cell viability values ≥ 80% were selected for uptake experiments (results presented in Fig. 3 and Table II). Monodansyl cadeverin (MDC), a clathrin-mediated endocytosis pathway inhibitor, was used at a concentration of 300 μM for cell studies. At this concentration, little toxicity was observed compared to the maximum tested concentration of 600 μM. Filipin (FIL), which inhibits the caveolae-mediated endocytosis pathway by binding to cholesterol inside caveolar pits and disrupting the pathway cycle, was used at a concentration of 4 μM for cell uptake experiments. Wortmannin (WORT), which inhibits the process of macropinocytosis, did not show a considerable level of toxicity at any of the tested concentrations (50 nM to 200 nM), hence 100 nM concentration was used for inhibition experiments as shown previously in literature (27). Dynasore (DYN), an inhibitor which is responsible for vesicle scission during both clathrin- and caveolin- mediated pathways and selectively inhibits dynamin 1 and dynamin 2 GTPases during uptake (8,26), showed increasing toxicity over the concentrations tested with an acceptable viability value at 50 μM, the value chosen for subsequent experiments.

Cytotoxicity of endocytosis inhibitors. Cell populations were incubated for 2 h at 37°C with various endocytosis inhibitors. The graph indicates the percentage of cell viability for the different concentrations for each endocytosis inhibitor and results are reported as Mean ± Standard Deviation with n = 6.
The literature shows that liposomes are predominantly endocytosed through either the clathrin- or caveolin- mediated endocytosis (29–31). Since liposomes prepared via microfluidics exhibit significantly decreased variability in size and polydispersity compared to those made through traditional processes, the present experiments are able to distinguish the uptake pathways experienced by each population of distinctly-sized liposomes and elucidate specific relationships between endocytic fate and vesicle size. The effect of vesicle size on cellular uptake was compared at different conditions of temperature (37°C and 4°C) and in the presence of each respective endocytosis pathway inhibitor at 37°C. Uptake of liposomes at 4°C represents passive cellular uptake: at this low temperature there is a global suppression of all active endocytotic pathways, thus any liposome uptake at 4°C is completely dependent on the ability of the particle to be passively internalized through the cell membrane. Conversely, the uptake of liposomes at 37°C in the absence of endocytosis inhibitors represents total cellular uptake when all available endocytosis mechanisms within Caco-2 cells may be fully utilized. In Fig. 4, uptake effect in presence of endocytosis inhibitors and at 4°C are all normalized to the total uptake at 37°C (which represents 100% total uptake in absence of endocytosis inhibitors). Therefore, any difference between the uptake at 37°C and the uptake under the influence of inhibitors represents the liposome uptake hampered by the inhibitor. However, the difference between the uptake of liposome in presence of endocytosis inhibitors and the uptake at 4°C represents the amount of liposomes not influenced by the pathway.

Liposome uptake in presence of endocytosis inhibitors. Liposomes of different sizes, 162.1 nm, 97.8 nm, 72.3 nm and 40.6 nm, were used for uptake studies in presence of endocytosis inhibitors. Data is normalized to the total uptake at 37°C (100%) and passive uptake at 4°C (starting point, 0%). The graph respresents the percentage of liposomes that are not affected by inhibitors, monodansyl cadeverin (MDC)(300 μM), wortmannin (WORT)(100 nM), filipin (FIL)(4 μM) and dynamin (DYN)(50 μM). All presented as Mean ± Standard Deviation with n = 4 (10,000 to 20,000 events each sample repeat ‘n’).
The presence of MDC (clathrin-mediated pathway inhibitor) caused a much greater inhibition of cellular uptake of 72.3 nm diameter liposome than compared to the other sizes, 162.1 nm, 97.8 nm and 40.6 nm (Fig. 4). Although cellular uptake of the larger liposomes appeared to be affected by MDC, when compared to the total uptake, an almost complete suppression of uptake was mainly observed for the medium sized liposomes (72.3 nm). Suggesting that the 72.3 nm liposomes may rely on the clathrin-dependent uptake mechanim for entry into Caco-2 cells. In contrast, MDC caused minimal effect on the uptake of the smallest liposomes (40.6 nm). These observations suggest that 40.6 nm diameter liposomes might be exploiting a clathrin-independent pathway for entry into Caco-2 cells.
The presence of WORT, (macropinocytosis uptake pathway inhibitor), the smaller liposomes (40.6 nm) experienced little change in their uptake as compared to the other sizes, suggesting that this pathway may not significantly influence their entry into Caco-2 cells. WORT pathway inhibitor like the MDC inhibitor, caused almost complete inhibition in cellular uptake of 72.3 nm liposomes (Fig. 4), suggesting that this size range may also be majorly influenced by the macropinocytosis uptake mechanism. The larger 162.1 nm and 97.8 nm liposomes were not very greatly influenced by this pathway in comparison to the other sizes.
Upon exposure to FIL (Fig. 4), (caveolar-mediated endocytosis inhibitor), there was very little inhibition in cellular uptake of the larger liposomes (97.8 nm and 162.1 nm), but an increased inhibition of small- and medium-sized liposomes (40.6 nm and 72.3 nm). These results suggest the possibility of small- and medium-sized liposomes following caveolae-mediated (clathrin-independent) endocytosis pathways, as opposed to their larger counterparts.
Under the presence of DYN (dynamin inhibitor), nearly complete inhibition of cellular uptake of small- and medium-sized liposomes (40.6 nm and 72.3 nm), whereas larger 97.8 nm and 162.1 nm diameter liposomes in comparison were not affected much by the inhibitor.
In conclusion, exposure to the combination of inhibitors of specific endocytosis pathways and liposomes of various sizes has been studied here to elucidate size-based uptake mechanisms experienced by Caco-2 cells in culture. When deriving a comparison between the effect of inhibitor on cellular uptake of liposomes of different sizes, the size ranges of 162.1 nm and 97.8 nm appeared to not be influenced by most of the tested pathway inhibitors. However when this comparison is made between the different inhibitor within the same size, for example with the 162.1 nm, the MDC and DYN has a slightly more effect on the uptake than compared to the FIL and WORT. This may suggest that this size could possibly be influenced by a clathrin- and dynamin- dependent endocytosis pathways. For the 97.8 nm the MDC seems to have the most effect compared to the other pathway inhibitors FIL, DYN and WORT, hence suggesting that the clathrin-dependent pathway may also be influencing this size range. All pathway inhibitors tested here influenced the uptake of 72.3 nm diameter liposomes indicating that liposomes of this size may have the ability to be taken up through several mechanisms within these cells. The smallest tested liposomes (40.6 nm) inhibited mainly by dependent by DYN indicating that the fate of particle internalization may depend primarily on a dynamin-dependent endocytosis pathway.
Intracellular Trafficking Using Confocal Microscopy
Intracellular trafficking of liposomes was observed in addition to determining their uptake mechanisms (Fig. 5). Liposomes were expected to co-localize with the endosomes and lysosomes over time. However the aim here was to see the differences in co-localization between the varied liposome sizes and endosome (EEA-1) or lysosome (LAMP-1) compartments after 1 h of incubation at 37°C. From Figs. 5 and 6, a trend was observed in the co-localization of liposomes, where the much smaller 40.6 nm and 72.3 nm liposomes co-localized with the endosomes slightly less than the 97.8 nm and 162.1 nm diameter liposomes. Although more than 40% of the 40.6 nm liposomes appear to have co-localized with endosomes, it may be possible that the remaining 60% may move further down the trafficking pathway, into late endosome or lysosome vesicles. Figures 5 and 7 shows that the 72.3 nm and 40.6 nm liposomes are more co-localized with the LAMP-1 lysosomal region than with EEA-1 endosomal regions. The 72.3 nm diameter liposomes co-localizing with the LAMP-1 regions much more compared to any of the other tested sizes, suggesting that a higher percentage of these diameter range liposomes might accumulate in the lysosomes and drug release may be quicker and more effective.

Co-localization of liposomes with EEA-1 and LAMP-1. Intracellular trafficking of liposomes was measured after 1 h incubation with Caco-2 cells. Co-localization between liposomes and endosomes (EEA1)/lysosomes (LAMP1) was measured using Mander’s overlap coefficient (Mx(oc)). Results are reported as Mean ± Standard Deviation where * represents a significant difference for (p < 0.05) by One-way ANOVA and by Tuckey’s Multiple Comparison test with n = 8.

Confocal imaging of co-localization of liposomes with endosomal regions. Caco-2 cells incubated for 1 h with DiI-C18 (red) fluorescently labeled liposomes, (a) 40.6 nm, (b) 72.3 nm, (c) 97.8 nm and (d) 162.1 nm liposomes. Endosomes were stained using AlexaFluor 488 (green) for anti EEA1 antibody. These samples were examined by confocal microscope (Nikon A1). The nucleus was stained with Dapi. The arrows in the merge image point to the co-localized regions. Scale bar is 5 μm.

Confocal imaging of co-localization of liposomes with lysosomal regions. Caco-2 cells incubated for 1 h with DiI-C18 (red) fluorescently labeled liposomes, (a) 40.6 nm, (b) 72.3 nm, (c) 97.8 nm and (d) 162.1 nm liposomes. Lysosomes were stained using AlexaFluor 488 (green) for anti Lamp-1 antibody. These samples were examined by confocal microscope (Nikon A1). The nucleus was stained with Dapi. Arrow in the merge images point to the co-localized regions . Scale bar is 10 μm.
Discussion
Microfluidic technology has enabled the production of nearly-monodisperse populations of liposomes with distinct diameters for size-specific cellular uptake studies. For our study liposomes were prepared using a method first demonstrated by Jahn, et al. (23), where solutions of nearly-monodispersed liposomes were achieved using hydrodynamic flow focusing. We adopted this method to prepare our liposomes for cellular uptake studies in which the relationships between endocytosis mechanisms and vesicle sizes were analyzed with high specificity. The ability to control liposome size and polydispersity, to an extent, has potential to have a large impact on numerous drug delivery applications and could improve upon currently available liposome-based drugs by rendering them more site-specific and efficient(13). In order to design effective liposomal drug delivery systems, it is critical to also understand the detailed mechanisms of their cellular trafficking and transport(12). In this work, liposomes prepared within microfluidic devices, via a hydrodynamic flow-focusing technique(23,28), ranged in mean diameters of 40.6 nm to 276.6 nm. The overall cellular uptake of liposomes over time (15 min and 60 min at 37°C) was observed to be dependent on liposome size in Caco-2 cells. Total cellular accumulation was higher for the smallest liposomes (40.6 nm) and for the medium range liposomes (72.3 nm) in comparison to the largest liposomes (276.6 nm) (Fig. 2). This illustrates the significant effect of size on cellular uptake, particularly that smaller liposomes are internalized more readily over time. When considering liposome based drug targeting for in vivo conditions the circulation, accumulation, in vivo drug release and clearance have been extensively studied revealing that amongst other things, size is an important factor when considering a vehicle for drug delivery(10,32). Harashima et al.(10), have shown a positive correlation to the effect of liposome size on the rate of clearance from blood and also suggesting that a decrease in the liposome size may reduce recognition by components in blood, like the mononuclear phagocytic system (MPS). Although in some cases liposome delivery and effectiveness is also dependent on the lipid composition (25,33,34) and surface charge or chemistry (35–37), it has been shown that size is also a major factor determining effectiveness of drug delivery. When considering liposome delivery for antitumor drugs, size appears to be a major factor affecting drug permeation into the in vivo tumor environment and local tumor tissue, where the optimal size has been shown to be in the range of ≤100 nm (10,32). This size range has also been shown to accumulate more within tumor tissues compared to much larger sizes (10). However, a liposome size of 100 nm may not always be optimal for all tumors, since tumor conditions such as the pore size of a given tumor’s vessels varies depending on the type of tumor, site of the growth, size of the tumor and the regression of the tumor site (38,39).
In this study we have demonstrated the in vitro effectiveness of smaller (40.6 nm to 72.3 nm) liposomes as compared to the much larger (97.8 nm – 276.6 nm range) liposomes on the uptake in Caco-2 cells, therefore suggesting that the former may be a more relevant size for specific targeting applications when translating this study to in vivo conditions. We also evaluated size-dependent liposome internalization by focusing on four main cellular uptake pathways, clathrin-mediated, caveolin-mediated, macropinocytosis and dynamin-dependent pathways for liposomes sizes of 40.6 nm to 162.1 nm. These endocytosis pathways are unique internalization mechanisms by which cells respond to certain micro and nanoparticles. The clathrin-mediated pathway is dependent on the formation of coated pits by the assembly of the protein clathrin, which forms a triskelion-like shape composed of three clathrin heavy chains and three light chains (14). The coated pit forms a vesicle upon endocytosis of particles. Vesicles formed through the clathrin-mediated pathway are typically about 120 nm to 150 nm in diameter (14,40,41). The caveolae-mediated pathway is dependent on the protein caveolin, which binds cholesterol to sphingolipids in certain areas and forms flask-like invaginations of the cell plasma membrane. Caveolae vesicles range from about 50 nm to 80 nm in diameter (14,41). The macropinocytosis pathway is a clathrin- and caveolin- independent, and actin-dependent process where the protrusions from the cell membrane collapse onto and fuse with the plasma membrane to generate large endocytic vesicles ranging from about 500 nm to 1,000 nm large (depending on the cell type) (14,41). This sort of pathway is not size specific and can internalize small to large particles. The dynamin-dependent pathway is an important receptor-mediated pathway in which the enzyme dynamin pinches vesicles from the plasma membrane during endocytosis (42,43). Some studies suggest that dynamin may also be involved in certain caveolae- and clathrin-independent pathways; for example, Ras homolog gene family member A (RhoA)-dependent pathway, cell division control protein 42 (Cdc42)-dependent pathway, and flotillin-dependent pathway (42). Studies have also suggest that dynamin may self-assemble into rings (typically range from 30 nm to 50 nm in diameter) which constrict invaginated coated pits around their necks to form budding coated vesicles (44). In our experiments we observed that the dynamin-dependent pathway specifically influenced the smaller 40.6 nm liposomes. Interestingly, 72.3 nm diameter liposomes also seemed to be affected by this pathway. Whereas the 97.8 nm to 162.1 nm liposomes were not affected as much by this pathway compared to the other smaller sizes. The larger, 97.8 nm to 162.1 nm, were not influenced by any of the pathways tested as much as the smaller 72.3 nm and 40.6 nm. The larger liposomes 162.3 nm seemed to be more affected by the MDC and DYN inhibitors than the FIL and WORT inhibitors, which may suggest that this size range may be influenced slightly by the clathrin- and dynamin- dependent pathways. Whereas the 97.8 nm liposomes were influenced by the MDC when compared to the other inhibitors FIL, WORT and DYN, thus suggesting that this size range may be influenced more by the clathrin-dependent pathway. In contrast, the 72.3 nm liposome seemed to be affected by all tested pathway inhibitors. Hence suggesting that this might be a useful size range for drug delivery since it utilizes most of the major trafficking pathways within cells. Since chemical inhibitors have a varied effectiveness in different cell lines and can somewhat be non-specific (26), hence liposome particles may behave differently depending on cell type and our results may be true for Caco-2 cellular uptake in specific. The intracellular trafficking experiments, using the confocal microscopy, shed further light on the environment that liposomes encounter after cellular uptake. Co-localization between the liposomes of varied size with endosomal and lysosomal markers was reported in Caco-2 cells. Early endosomal accumulation of 97.8 nm diameter liposomes was much greater than lysosomal accumulation after 1 h of incubation. For the 72.3 nm liposomes after 1 h incubation they seemed to be more accumulated within lysosomal regions than compared to endosomal regions. A similar trend was observed for the 40.6 nm diameter liposomes where there was slightly higher lysosomal accumulation of these liposomes compared to the endosomal regions. Therefore a greater amount of the 40.6 nm and 72.3 nm diameter liposomes travel to the lysosome for degradation, which is a key step in the effective targeting and drug release (14). Thus, we have shown that total liposome uptake is dependent on size and that there are several mechanisms that come into play during the uptake process that may also be influenced by size.
Previous studies probing the cellular uptake and trafficking of liposomes have been based on populations of vesicles with high levels of poly-dispersity due to the limitations of contemporary synthesis methods (8,28), thus the ability to highlight the true fate of each individual vesicle size within the range of interest was limited. Microfluidic mixing of chemical species provides the unique ability to produce nearly-monodisperse populations of liposomes with distinct, varying size, this is a highly advantageous quality for increasing specificity and efficacy while reducing toxicity of liposomes for drug delivery applications. Microfluidic liposome preparation also uses much less quantities of solution and may be a more cost effective method for liposome production(28) (problems concerning expenses for academia and industry mentioned in a review by Barenholz, et al. (45)). The ability to synthesize homogenous populations of liposomes also permits a higher level of control over the intracellular fate of the drug carrier, thus aiding in more sophisticated design of liposomal drug delivery systems.
Conclusions
Here we reported a detailed study of cellular uptake mechanisms and intracellular trafficking related to the uptake of liposome populations prepared by a novel microfluidic flow focusing technique described by Jahn et al.(28). The unique ability of microfluidic flow focusing to produce nearly-monodispersed populations of distinct sizes is highly advantageous for increasing the specificity for drug delivery applications. We found that the liposome uptake is size dependent and increases with a decrease in diameter. The mechanisms involved in the uptake of liposomes are also dependent on the sizes, where the smaller 40.6 nm liposomes seem to depend on a dynamin dominant pathway. However the slightly larger diameter liposomes (97.8 nm to 162.1 nm), when compared to the smaller sizes, did not show any significant dependency on any particular uptake pathways that were tested, but when the larger liposomes were compared among themselves they showed slight dependency on the clathrin-mediated pathway. The uniqueness observed in this study was the intermediate size range of 72.3 nm that seemed to be dependent on almost all the tested mechanisms. Also this size range showed the most acculmulation within the lysosomal regions compared to the other tested liposome diameters. In this study we found that size plays a big role in facilitating the recognition of particles by a particular mechanism. Knowledge of detailed mechanisms of liposome sizes was better facilitated with the use of microfluidic flow focusing and this may assist in designing better liposome populations for drug delivery for various applications in the future.
Abbreviations
- AF4 :
-
assymetric flow field-flow fractionation
- BSA:
-
bovine serum albumin
- Caco-2:
-
cancer colon (human colorectal adenocarcinoma cells)
- DAPI:
-
4’,6-diamino-2-phenylindole
- DiI C18 :
-
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- DMEM:
-
Dulbecco’s modified eagle’s medium
- DPBS:
-
Dulbecco’s phosphate buffer saline
- DPPC:
-
Dipalmitoylphosphatidylcholine
- DYN:
-
dynasore
- EDTA:
-
ethylenediamine tetra acetic acid
- EEA-1:
-
early endosome antigen -1
- FIL:
-
filipin
- FRR:
-
flow rate ratio
- HBSS:
-
Hank’s balanced salt solution
- LAMP-1:
-
lysosome-associated membrane protein - 1
- MALLS:
-
multiangle laser light scattering
- MDC:
-
monodansyl cadaverine
- PBS:
-
phosphate buffer saline
- PDI:
-
poly dispersity index
- PDMS:
-
poly (dimethylsiloxane)
- PEG:
-
poly (ethylen glycol)
- PEG2000-PE:
-
dipalmitoylphosphatidylethanolamine-poly(ethylene glycol) 2000
- QELS:
-
quasi-elastic light scattering
- WORT:
-
wortmannin
- WST-1:
-
water-soluble tetrazolium salt
References
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–60.
Allen TM. Liposomes - opportunities in drug delivery. Drugs. 1997;54(Supplement 4):8–14.
Storm G, Crommelin DJ. Liposomes: quo vadis? Pharm Sci Technol Today. 1998;1(1):19–31.
Lasic DD. Novel applications of liposomes. Trends Biotechnol. 1998;16(7):307–21.
Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–36.
Barenholz Y. Doxil®--the first FDA-approved nano-drug: lessons learned. J Control Release: Off J Control Release Soc. 2012;160(2):117–34. Elsevier B.V.
Gregoriadis G. Engineering liposomes for drug delivery: progress and problems. Trends Biotechnol. 1995;13(12):527–37.
Edwards KA, Baeumner AJ. Liposomes in analyses. Talanta. 2006;68(5):1421–31.
Litzinger DC, Buiting AMJ, Van Rooijen N, Huang L. Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. Biochim Biophys Acta (BBA) Biomembr. 1994;1190(1):99–107.
Nagayasu A, Uchiyama K, Kiwada H. The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv Drug Deliv Rev. 1999;40(1–2):75–87.
Lee R, Low P. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J Biol Chem. 1994;259(5):3196–204.
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–27. Nature Publishing Group.
Wang J, Byrne JD, Napier ME, DeSimone JM. More effective nanomedicines through particle design. Small. 2011;7(14):1919–31. WILEY-VCH Verlag.
Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol life Sci: CMLS. 2009;66(17):2873–96.
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science (New York, NY). 2004;303(5665):1818–22.
Wagner A, Vorauer-Uhl K, Kreismayr G, Katinger H. The crossflow injection technique: an improvement of the ethanol injection method. J Liposome Res. 2002;12(3):259–70.
Olson F, Hunt CA, Szoka FC, Vail WJ, Papahadjopoulos D. Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim Biophys Acta (BBA) Biomembr. 1979;557(1):9–23.
Szoka F Jr, Papahadjopoulos D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu Rev Biophy. 1980;9:467–508.
Batzri S, Korn ED. Single bilayer liposomes prepared without sonication. Biochim Biophys Acta (BBA) Biomembr. 1973;298(4):1015–9.
Kremer J. Vesicles of variable diameter prepared by a modified injection method. Biochemistry. 1977;16(17):3932–5.
Jahn A, Stavis SM, Hong JS, Vreeland WN, DeVoe DL, Gaitan M. Microfluidic mixing and the formation of nanoscale lipid vesicles. ACS Nano. 2010;4(4):2077–87.
Jahn A, Reiner JE, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Preparation of nanoparticles by continuous-flow microfluidics. J Nanopart Res. 2008;10(6):925–34.
Jahn A, Vreeland WN, Gaitan M, Locascio LE. Controlled vesicle self-assembly in microfluidic channels with hydrodynamic focusing. J Am Chem Soc. 2004;126(9):2674–5.
Hood R, Shao C, Omiatek D, Vreeland W, DeVoe D. Microfluidic synthesis of PEG- and folate-conjugated liposomes for one-step formation of targeted stealth nanocarriers. Pharm Res . Springer US; 2013;1–11.
Allen TM, Austin GA, Chonn A, Lin L, Lee KC. Uptake of liposomes by cultured mouse bone marrow macrophages: influence of liposome composition and size. Biochim Biophys Acta. 1991;1061(1):56–64.
Goldberg DS, Ghandehari H, Swaan PW. Cellular entry of G3.5 poly (amido amine) dendrimers by clathrin- and dynamin-dependent endocytosis promotes tight junctional opening in intestinal epithelia. Pharm Res. 2010;27(8):1547–57.
Pollock S, Antrobus R, Newton L, Kampa B, Rossa J, Latham S, et al. Uptake and trafficking of liposomes to the endoplasmic reticulum. FASEB J: Off Publ Fed Am Soc Exp Biol. 2010;24(6):1866–78.
Jahn A, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Microfluidic directed formation of liposomes of controlled size. Langmuir: ACS J Surf Colloids. 2007;23(11):6289–93.
Straubinger RM, Hong K, Friend DS, Papahadjopoulos D. Endocytosis of liposomes and intracellular fate of encapsulated molecules: encounter with a low pH compartment after internalization in coated vesicles. Cell. 1983;32(4):1069–79.
Kamps J, Scherphof G. Receptor versus non-receptor mediated clearance of liposomes. Adv Drug Deliv Rev. 1998;32(1–2):81–97.
Hong K, Yoshimura T, Papahadjopoulos D. Interaction of clathrin with liposomes: pH-dependent fusion of phospholipid membranes induced by clathrin. FEBS Lett. 1985;191(1):17–23.
Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharm Rev. 1999;51(4):691–743.
Barza M, Stuart M, Szoka F. Effect of size and lipid composition on the pharmacokinetics of intravitreal liposomes. Invest Ophthalmol Vis Sci. 1987;28(5):893–900.
Mayer LD, Tai LC, Ko DS, Masin D, Ginsberg RS, Cullis PR, et al. Influence of vesicle size, lipid composition, and drug-to-lipid ratio on the biological activity of liposomal doxorubicin in mice. Cancer Res. 1989;49(21):5922–30.
Souhami RL, Patel HM, Ryman BE. The effect of reticuloendothelial blockade on the blood clearance and tissue distribution of liposomes. Biochim Biophys Acta (BBA) Gen Subj. 1981;674(3):354–71.
Lee R, Low P. Folate-mediated tumor cell targetting of liposome-entrapped doxorubicin. Biochim Biophys Acta (BBA) Biomembr. 1995;1233:134–44.
Koren E, Apte A, Jani A, Torchilin VP. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release: Off J Control Release Soc. 2012;160(2):264–73. Elsevier B.V.
Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–12.
Hirano A, Matsui T. Vascular structures in brain tumors. Human Pathol. 1975;6(5):611–21.
Bareford L, Swaan P. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–58.
Wang J, Byrne J, Napier M, DeSimone J. More effective nanomedicines through particle design. Small. 2011;7(14):1919–31.
Sever S. Dynamin and endocytosis. Curr Opin Cell Biol. 2002;14:463–7.
Gu C, Yaddanapudi S, Weins A, Osborn T, Reiser J, Pollak M, et al. Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J. 2010;29(21):3593–606. Nature Publishing Group.
Hinshaw JE, Schmid SL. Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature. 1995. p. 190–2.
Barenholz Y. Liposome application: problems and prospects. Curr Opin Colloid Interface Sci. 2001;6(1):66–77.
Acknowledgments and Disclosures
Certain commercial equipment, instruments, or materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Financial support was provided by the Department of Energy (DOE), grant number DE-FG02-08CH11527. Cathy Storer, B.S., Regina Harley, M.S. and Marcelo Sztein, M.D. from the Flow Cytometry Core Facilities, Centre for Vaccine Development, School of Medicine, University of Maryland, Baltimore, USA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Abhay U. Andar and Renee R. Hood contributed equally to this work.
Rights and permissions
About this article
Cite this article
Andar, A.U., Hood, R.R., Vreeland, W.N. et al. Microfluidic Preparation of Liposomes to Determine Particle Size Influence on Cellular Uptake Mechanisms. Pharm Res 31, 401–413 (2014). https://doi.org/10.1007/s11095-013-1171-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-013-1171-8