
Abstract
We calculate the different geometric isomers of spin clusters composed of a small number of alkali-metal atoms using the UB3LYP density-functional method. The electron density distribution of clusters changes according to the value of total spin. Steric structures as well as planar structures arise when the number of atoms increases. The lowest spin state is the most stable and Li n , Na n , K n , Rb n , and Cs n with n = 2–8 can be formed in higher spin states. In the highest spin state, the preparation of clusters depends on the kind and the number of constituent atoms. The interaction energy between alkali-metal atoms and rare-gas atoms is smaller than the binding energy of spin clusters. Consequently, it is possible to self-organize the alkali-metal-atom clusters on a non-wetting substrate coated with rare-gas atoms.
Similar content being viewed by others
Introduction
Spin clusters are expected to be used as basic functional elements of nano-magnetic and quantum systems (Barth et al. 2005) employed for ultrahigh-density magnetic recording (Gambardella 2006; Affronte 2009) and quantum computing (Meier et al. 2003). Although the spin clusters have been observed in condensed matter or polymer complex (Lee et al. 2002), nothing more than trimers have been formed in an isolated system. One of the most fundamental spin clusters is made up of a small number of atoms. Indeed, we are planning to create the spin clusters by self-organization of alkali-metal atoms. In this scheme, spin-polarized cold atoms produced by a magneto-optical trap followed by optical pumping are slowly deposited on a substrate coated with rare-gas atoms (Torikai 2000). Thanks to the non-wetting interaction between alkali-metal atoms and rare-gas atoms, spin clusters are considered to be formed on the substrate.
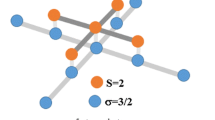
A lot of studies on spin clusters have just addressed the most stable state, i.e., the lowest spin state, but had less interest in metastable states with higher spin. Note that the shape of atomic clusters strongly depends on the spin state of each atom. Figure 1 shows the case of Rb4 as an example. The four spin-polarized Rb atoms form a parallelogram cluster when S = 0, while a square cluster when S = 1. The different geometries of Cs n clusters with n = 2–8 have been calculated with Gaussian 98 (Srinivas et al. 2007). The density-functional study with the BPW91 functional (Becke 1988; Perdew and Wang 1992) indicates that the coupling in the highest spin state is impossible even in the dimer. It is not true because the dimer with S = 1 is experimentally observed (Fioretti et al. 1998). The error is due to the use of a less-accurate expression of the exchange-correlation potential. To improve simulations with respect to higher spin states, we use the UB3LYP functional involving a more accurate formula of the exchange-correlation potential (Lee et al. 1988). In this paper, the characteristics of alkali-metal-atom spin clusters are systematically examined for all stable isotopes of Li, Na, K, Rb, and Cs using Gaussian 03 (Frisch et al. 2003) associated with its graphical user interface GaussView 03.

Self-organization of four Rb atoms under spin polarization. Each Rb atom has either up-spin or down-spin. The motif of the cluster is parallelogram in the singlet (S = 0) state, while square in the triplet (S = 1) state, where S is the total spin of the system. The blue cloud drawn on the right-hand side represents the density distribution of the peripheral electrons. (Color figure online)
Computational approach
Spin-exchange and correlation interactions are considered on the basis of the typical density-functional theory. Under the local density approximation and the generalized gradient approximation, the exchange and correlation energies are expressed as a functional of both the density function and its gradient at a point in space (Parr et al. 1989). The UB3LYP hybrid functional, which consists of three exchange terms coupled by Becke’s three parameters and the Lee-Yang-Parr correlation term (Lee et al. 1988; Mielich et al. 1989; Becke 1993) is employed for conducting harmonic vibrational calculations and determining an optimal motif of each cluster. For Li n , Na n , and K n , all-electron calculations are performed using the 6-311+G(3df) basis set with the largest triply split-valence with polarization functions. On the other hand, for Rb n and Cs n composed of heavier atoms, the peripheral-electron calculations are performed using the LANL2DZ basis set, where the contribution of inner electrons is approximated by double-\(\zeta\)-type effective core potentials (Handy et al. 1984; Wadt and Hay 1985; Nicklass et al. 1995).
In the following calculations, we consider from dimers to octamers that consist of the most abundant atomic species among stable isotopes, i.e., 7Li n , 23Na n , 39K n , 85Rb n , and 133Cs n with n = 2–8. We also examined 6Li n , 40K n , 41K n , and 87Rb n . As a result, the motif, the binding energy, and the bond length, they are discussed below, were almost the same among the clusters composed of a different isotope of the same atomic species. So, we simply denote 7Li n , 23Na n , 39K n , 85Rb n , and 133Cs n just as Li n , Na n , K n , Rb n , and Cs n , and will describe about these essential clusters hereafter.
To confirm that the UB3LYP functional is the best choice, we also performed the calculations of alkali-metal-atom clusters using unrestricted Hartree-Fock (UHF), unrestricted second-order perturbed Møller-Plesset (UMP2), and unrestricted Becke-3 with Perdew-Wang 91 (UB3PW91) (Becke 1993; Perdew et al. 1996). Table 1 shows the binding energy of dimers with S = 0 obtained from these methods in comparison to the experimental data (Zuchowski et al. 2010). Here, the 6-311+G(3df) basis set is used for Li2, Na2, and K2, and the LANL2DZ basis set is used for Rb2 and Cs2. As shown in Fig. 2, the UB3LYP method gives the closest values to the experimental ones. Similarly, the bond length and the ionization energy obtained with the UB3LYP method are also in good agreement with the experimental results (Florez et al. 2009). Incidentally, the UBLYP functional used in Florez et al. (2009) gives better values with respect to the ionization potential. On the other hand, the UB3LYP functional gives better values with respect to the binding energy of electrically-neutral clusters.

Comparison of the binding energy \(\Updelta\) of Li2, Na2, K2, Rb2, and Cs2 in the lowest spin state estimated by means of different theoretical methods with the experimental data. The black square, the red circle, the green up-triangle, the blue down-triangle, and the light-blue diamond indicate UHF, UMP2, UB3PW91, UB3LYP, and the experimental data, respectively. (Color figure online)
Cluster geometry
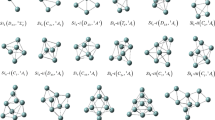
We calculated the geometry of various isomers with different spin S. The spin density function \(\rho_{\alpha}(\user2{r})\) of the α-electrons occupying the highest orbital is drawn as a blue cloud for each cluster motif in Figs. 3, 4, 5, 6, 7, and 8. In addition, the spin density function \(\rho_{\beta}(\user2{r})\) of the β-electrons occupying the highest orbital is drawn as a red cloud provided that the distribution is different from that of the α-electrons, i.e., except for S = 0 and the highest spin state. Note that the middle small sphere expresses nucleus. For Li n , Na n , and K n obtained from all-electron calculations, the core electron distribution is also shown with dark blue or dark red. When exchanging α-electrons for β-electrons, the cases giving the same results are removed. In general, the motif of spin clusters is planar (2D) for n = 2 and 3, and the steric (3D) structures as well as the planar ones arise for n ≥ 4. The most stable state is the lowest spin state, i.e., singlet (S = 0) for clusters with even numbers of atoms and doublet (S = 1/2) for clusters with odd numbers of atoms. The density cloud visualizes the strength of binding. When the distribution of each electron overlaps considerably, the cluster is stable. Otherwise, it is unstable.

Motifs and α-electron density distributions of dimers
Figure 3 shows the case of dimers. In the triplet (S = 1) state with parallel spin, Li2 can be formed. It has a wide electron distribution, compared to the singlet state, because of repulsion between electrons. The preparation of Na2 and Cs2 with S = 1 is critical since the binding energy is very small, as can be noted from the α-electron cloud.
Figure 4 shows the two cases of (a) trimers and (b) tetramers. The most stable structure with S = 1/2 is triangle for Li3 and Na3, but normal chain for other trimers. The density of α-electron is large at the both ends of clusters as seen in the blue cloud, while that of β-electron is large at the center as seen in the red cloud, or vice versa In the quartet (S = 3/2) state, the motif is equilateral triangle. Meanwhile, as to tetramers, the most stable structure with S = 0 is parallelogram. In the triplet state, Li4, Na4, and K4 are rhombus, but Rb4 and Cs4 are square. In the quintet (S = 2) state, rhombus tetramers except Rb4 can be formed, although Na4, K4, and Cs4 are weakly-bound. The highest spin tetramers have a spatially-extended electron distribution and form a poor bound. Consequently, it is advantageous to be steric in the highest spin state. Indeed, the regular tetrahedral structure arises, which is one of the closest packed structures.

Motifs and α- and β-electron density distributions of a trimers and b tetramers
When n ≥ 5, steric structures appear in each spin state. As shown in Fig. 5a, the planar structure of pentamers is basically edge-capped rhombus, but K5 with S = 3/2 gets distorted. In the sextet (S = 5/2) state, Na5 and Rb5 cannot be formed. On the other hand, as shown in Fig. 5b, the steric structure of pentamers is approximately square pyramid, although the bottom is not always plane.

Motifs and α- and β-electron density distribution of pentamers, where a the 2D case and b the 3D case
Figure 6 shows (a) the 2D case and (b) the 3D case of hexamers. There are four types of planar structures: planar pyramid in the lowest spin state, and parallelogram-arranged, pentagon, or square-arranged shapes in higher spin states. In the septet (S = 3) state, Li6 is pentagon, Na6 and Cs6 are parallelogram-arranged, but K6 and Rb6 cannot be formed. Meanwhile, there are three types of steric structures: pentagonal pyramid in the lowest spin state, and capped trigonal bipyramid or octahedron in higher spin state. In the highest spin state, Li6, Na6, and K6 are capped trigonal bipyramid, but Rb6 and Cs6 are octahedron. As shown in Fig. 6b, the shape of cluster with the highest spin is quite different from that with the lowest spin.

Motifs and α- and β-electron density distribution of hexamers, where a the 2D case and b the 3D case
Figure 7 shows (a) the 2D case and (b) the 3D case of heptamers. The planar structure is trapezoidal-arranged for Li7 and hexagon for other clusters. In the octet (S = 7/2) state, Na7, K7, and Rb7 cannot be formed. In contrast, the most stable steric structure is decahedron. The bicapped trigonal bipyramid, capped octahedron, and capped tetragonal bipyramid also arise in higher spin states. Only Na7 cannot be formed in the highest spin state.

Motifs and α- and β-electron density distribution of heptamers, where a the 2D case and b the 3D case
Figure 8 shows (a) the 2D case and (b) the 3D case of octamers. In the highest spin state, only Na8 cannot be formed for both the 2D and 3D cases. The most stable steric structure is divided into two motifs of parallelogram-arranged and edge-capped hexagon, differently from the previous cases.

Motifs and α- and β-electron density distribution of octamers, where a the 2D case and b the 3D case
Binding energies
The binding energy of a spin cluster is defined as
where \(\epsilon_{0}\) is the energy of each atom and \(E^{(n)}=\sum_{i}\epsilon_{i}^{(n)}\) is the total electron energy of the cluster with the number n of atoms.
Figure 9 shows the binding energy per atom \(\Updelta/n\) of (a) Li n , (b) Na n , (c) K n , (d) Rb n , and (e) Cs n for each spin. If both planar and steric structures are possible, a higher energy is marked. As seen in these figures, \(\Updelta/n\) is larger as the number n increases, while it is smaller as the spin S increases. The most stable state is S = 0 when n is even and S = 1/2 when n is odd. The binding energy per atom of the singlet state increases from 0.44 eV for Li2 to 0.83 eV for Li8, from 0.37 eV for Na2 to 0.57 eV for Na8, from 0.25 eV for K2 to 0.41 eV for K8, from 0.22 eV for Rb2 to 0.33 eV for Rb8, and from 0.19 eV for Cs2 to 0.29 eV for Cs8. The highest spin state can be formed except for Na7, Na8, K2, and Rb2. The strength of binding remains almost unchanged even if the constituent isotope is different such as 6Li n and 7Li n . In contrast, it weakens as the mass of the constituent atom increases, for example, \(\Updelta/n\) of K n is smaller than that of Li n .

Binding energy per atom \(\Updelta/n\) of (a) Li n , (b) Na n , (c) K n , (d) Rb n , and (e) Cs n for each spin state. The black square indicates S = 0 and S = 1/2, the red circle indicates S = 1 and S = 3/2, the green up-triangle indicates S = 2 and S = 5/2, the blue down-triangle indicates S = 3 and S = 7/2, and the light-blue diamonds indicates S = 4. (Color figure online)
The steric structures arise in the cases of n ≥ 5 with the lowest spin. Figure 10 shows the 2D and 3D binding energies per atom \(\Updelta/n\) of (a) Li n , (b) Na n , (c) K n , (d) Rb n , and (e) Cs n in the lowest spin state. The black square and the red open square indicate the 2D case and the 3D case, respectively. For Li n , the steric structure is more stable than the planar one. For other clusters, the planar structure is more stable than the steric one below n = 6, but the steric structure is more stable than the planar one over n = 6. As shown in Fig. 10, the 2D binding energy decreases at n = 3 except for lithium and increases again until n = 6. This implies the well-known fact that the dimers and planar hexamers of alkali-metal atoms are comparatively stable.

Binding energy per atom \(\Updelta/n\) of a Li n , b Na n , c K n , d Rb n , and e Cs n in the lowest spin state. The black square and the red open square indicate the 2D and 3D cases, respectively. (Color figure online)
The clusters composed of spin-polarized alkali-metal atoms have ferromagnetic properties (Duan et al. 2007). Figure 11 shows the binding energy per atom \(\Updelta/n\) of (a) Li n , (b) Na n , (c) K n , (d) Rb n , and (e) Cs n in the highest spin state. The black circle and the red open circle indicate the 2D case and the 3D case, respectively. The steric structure arises in the case of n ≥ 4 except for sodium. The steric Li n is most likely to be formed and the binding energy is almost ten times as much as that of K n . The planar Na n can be formed when n = 2, 3, 4, and 6, while the steric Na n cannot be formed except for n = 4 and 5. The planar K n cannot be formed when n = 2, 6, and 7. The planar Rb n can be formed when n = 3 and 8. The preparation of Cs n is similar to Li n , but the binding energy is approximately a twentieth.

Binding energy per atom \(\Updelta/n\) of a Li n , b Na n , c K n , d Rb n , and e Cs n in the highest spin state. The black circle and the red open circle indicate the 2D and 3D cases, respectively. (Color figure online)
Bond length
Next, we estimated the size of each cluster. Figure 12 shows the longest bond length of (a) Li n , (b) Na n , (c) K n , (d) Rb n , and (e) Cs n for both 2D and 3D cases in the lowest spin state. The bond length does not make much difference on the number n of atoms whether the structure is planar or steric, although the latter is slightly longer than the former. The longest bond length is between 2.7 and 3.1 Å for Li n , between 3.0 and 3.8 Å for Na n , between 3.9 and 4.6 Å for K n , between 4.2 and 4.7 Å for Rb n , and between 4.7 and 5.2 Å for Cs n .

Bond length of a Li n , b Na n , c K n , d Rb n , and e Cs n in the lowest spin state. The black square and the red open square indicate the 2D and 3D cases, respectively. (Color figure online)
Figure 13 shows the longest bond length of (a) Li n , (b) Na n , (c) K n , (d) Rb n , and (e) Cs n for both 2D and 3D cases in the highest spin state. The value is between 3.4 and 3.8 Å for Li n , between 9.4 and 11.6 Å for Na n , between 5.4 and 5.5 Å for K n , between 6.8 and 7.4 Å for Rb n , and between 7.2 and 7.5 Å for Cs n . The bond length in the highest spin state is longer than that in the lowest spin state.

Bond length of a Li n , b Na n , c K n , d Rb n , and e Cs n in the highest spin state. The black circle and the red open circle indicate the 2D and 3D cases, respectively. (Color figure online)
Discussions
Table 2 summarizes the feasibility of preparing the total spin-polarized clusters. The filled circle indicates that a planar structure can be formed, while the open circle indicates that a steric structure can be formed. The double circle indicates that both planar and steric structures can be formed. The cross indicates that a cluster cannot be formed.
Notice that Li n with n ≥ 2 can be formed in the highest spin state. The binding energy is larger than that of clusters composed of the other alkali-metal atoms. Also, Cs n with n ≥ 2 can be formed in the highest spin state, but it is unstable since the binding energy is considerably small. On the other hand, K2 and Rb2 cannot be formed in the highest spin state due to Pauli repulsion. Sodium cannot form heptamer and octamer in the highest spin state, although the reason is unclear from the current calculations.
We are advancing an experiment of producing the spin clusters on a substrate. Figure 14 schematically shows the configuration. First, alkali-metal atoms cooled below 100 μK are accumulated in the center of a magneto-optical trap (MOT) with three pairs of two oncoming σ+–σ− light beams, which are at right to one another, and a pair of anti-Helmholtz coils (Raab et al. 1987). Next, the cold atoms are released by turning off MOT. Then, the freely falling atoms are spin-polarized by illuminating with pumping light beams, where Zeeman splitting is made by applying a static magnetic field with an additional pair of Helmholtz coils. There are two kinds of optical pumping (Happer 1972). The spin alignment is performed with a linearly-polarized light beam perpendicular to the static magnetic field (π-pumping) for generation of the S = 0 lowest spin state. On the other hand, the spin orientation is performed with a circularly-polarized light beam parallel to the static magnetic field (σ-pumping) for the generation of the highest spin state. Other intermediate spin states can be generated using several pumping light beams (Wang et al. 2007). We will report the spin-polarization experiment elsewhere.

Sketch of the experimental configuration for preparation of spin clusters on a substrate coated with rare-gas atoms. Cold alkali-metal atoms falling from a magneto-optical trap are spin-polarized by light beams for optical pumping under a static magnetic field
The interaction between an atom and a substrate is usually stronger than that between atoms. To prevent the spin-polarized cold atoms from interacting with the substrate, we coat the surface of the substrate with rare-gas atoms. It is because alkali-metal atoms are expected to be non-wetting against rare-gas atoms (Cahn 1977). Table 3 shows the theoretical value of interaction between alkali-metal atoms and rare-gas atoms in comparison to the experimental data (Goll et al. 2006). The best choice is argon since the interaction is least. Lithium and potassium clusters can be generated on the substrate coated with argon atoms in every spin state. Similarly, other alkali-metal atom clusters can be organized except for the highest spin state. The steric sodium tetramer can be exceptionally formed in the highest spin state. The preparation of the steric rubidium tetramer and octamer can be possible even in the highest spin state. The total spin-polarized cesium clusters will also glow up on the argon-coated substrate except for dimer.
As will be appreciated from the bond length, the size of a spin cluster is several angstrom. Accordingly, the spin clusters can be used as material for ultrahigh-density recording exceeding 1 bit/nm2, which approximately equals 1 Pbit/inch2, based on, for example, the structure change. Figure 15 shows the case of Rb4. The binding energy of the parallelogram structure in the lowest spin state is 0.94 eV and the value is smaller than the photon energy 1.59 eV of the Rb D2 line with the wavelength of 780.2 nm in vacuum. Consequently, the parallelogram Rb4 with S = 0 will change to the square Rb4 with S = 1 by absorbing a σ+-photon. Near-field light is expected to be used for such local-selective interaction.

Structure change of Rb4-dependent on spin arrangement. The shape is parallelogram when S = 0, square when S = 1, and regular tetrahedron when S = 2
Conclusion
We determined the motif and the electron density distribution of spin clusters composed of alkali-metal atoms using the UB3LYP hybrid functional. The shape strongly depends on the total spin S of the system. The 2D structure is dominant in the case where the number n of constituent atoms is small, but the 3D structure is gradually preferable as n increases. In the lowest spin state which is most stable, only 2D structures are formed when n ≤ 4 and 3D structures also arise when n ≥ 5. Meanwhile, in the highest spin state, 3D structures arise when n ≥ 4.
It is possible to prepare a cluster of all species in each spin state except for the highest spin state. The binding energy peaks when S = 0 for even n and S = 1/2 for odd n. It is proportional to n, but inversely proportional to S. Lithium clusters have the largest binding energy among alkali-metal atom clusters in the highest spin state. Cesium clusters can be also formed, although the binding energy is about a twentieth compared to the lithium clusters. Concerning sodium, potassium, and rubidium, preparation of a cluster depends on n. The bond length is independent of n and almost constant, but gets longer with S.
The binding energy is larger than the interaction energy between alkali-metal atoms and rare-gas atoms. It allows us to create spin clusters on a substrate covered with rare-gas atoms. The demonstration experiment with an argon-coated substrate is in progress.
References
Affronte M (2009) Molecular nanomagnets for information technologies. J Mater Chem 19:1731–1737. doi:10.1039/b809251f
Barth JV, Costantini G, Kern K (2005) Engineering atomic and molecular nanostructures at surfaces. Nature 437:671–679. doi:10.1038/nature04166
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098–3100. doi:10.1103/PhysRevA.38.3098
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652. doi:10.1063/1.464913
Cahn JW (1977) Critical point wetting. J Chem Phys 66:3667–3672. doi:10.1063/1.434402
Duan TC, Nakano T, Nozue Y (2007) Magnetic and optical properties of Rb and Cs clusters incorporated into Zeolite A. e-J Surf Sci Nanotech 5:6–11
Fioretti A, Comparat D, Crubellier A, Dulieu O, Masnou-Seeuws F, Pillet P (1998) Formation of cold Cs2 molecules through photoassociation. Phys Rev Lett 80:4402–4405. doi:10.1103/PhysRevLett.80.4402
Florez E, Fuentealba P (2009) A theoretical study of alkali metal atomic clusters: from Li n to Cs n (n = 2 − 8). Int J Quant Chem 109:1080–1093. doi:10.1002/qua.21906
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Rob MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03. Gaussian Inc., Wallingford
Gambardella P (2006) Magnetic nanostructures: quantum chains with a spin. Nat Mater 5:431–432. doi:10.1038/nmat1662
Goll E, Werner H-J, Stoll H, Leininger T, Gori-Giorgi P, Savin A (2006) A short-range gradient-corrected spin density functional in combination with long-range coupled-cluster methods: application to alkali-metal rare-gas dimers. Chem Phys 329:276–282. doi:10.1016/j.chemphys.2006.05.020
Handy NC, Schaefer HF (1984) On the evaluation of analytic energy derivatives for correlated wave functions. J Chem Phys 81:5031–5033. doi:10.1063/1.447489
Happer W (1972) Optical pumping. Rev Mod Phys 44:169–249. doi:10.1103/RevModPhys.44.169
Lee S-H, Broholm C, Ratcliff W, Gasparovic G, Huang Q, Kim TH, Cheong S-W (2002) Emergent excitations in a geometrically frustrated magnet. Nature 418:856–858. doi:10.1038/nature00964
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789. doi:10.1103/PhysRevB.37.785
Meier F, Levy J, Loss D (2003) Quantum computing with spin cluster qubits. Phys Rev Lett 90:047901. doi:10.1103/PhysRevLett.90.047901
Mielich B, Savin A, Stoll H, Preuss H (1989) Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem Phys Lett 157:200–206. doi:10.1016/0009-2614(89)87234-3
Nicklass A, Dolg M, Stoll H, Preuss H (1995) Ab initio energyadjusted pseudopotentials for the noble gases Ne through Xe: calculation of atomic dipole and quadrupole polarizabilities. J Chem Phys 102:8942–8952. doi:10.1063/1.468948
Parr RG, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, New York
Perdew JP, Burke K, Wang Y (1996) Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys Rev B 54:16533–16539. doi:10.1103/PhysRevB.54.16533
Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B 45:13244–13249. doi:10.1103/PhysRevB.45.13244
Raab E, Prentiss M, Cable A, Chu S, Pritchard D (1987) Trapping of neutral-sodium atoms with radiation pressure. Phys Rev Lett 59:2631–2634. doi:10.1103/PhysRevLett.59.2631
Srinivas S, Torikai E (2007) A density-functional study of the structure and self-organization in spin clusters. J Magn Magn Mater 310:2390–2392. doi:10.1016/j.jmmm.2006.10.747
Torikai E (2000) Study of the self-organization of spin-polarized atomic clusters on a solid rare gas surface. RIKEN Rev 27:82–85
Wadt WR, Hay PJ (1985) Ab initio effective core potentials for molecular calculations—potentials for main group elements Na to Bi. J Chem Phys 82:284–298. doi:10.1063/1.448800
Wang B, Han Y, Xiao J, Yang X, Zhang C, Wang H, Xiao M, Peng K (2007) Preparation and determination of spin-polarized states in multi-Zeeman-sublevel atoms. Phys Rev A 75:051801. doi:10.1103/PhysRevA.75.051801
Zuchowski PS, Hutson JM (2010) Reactions of ultracold alkali-metal dimers. Phys Rev A 81:060703. doi:10.1103/PhysRevA.81.060703
Acknowledgments
We thank Prof. Sudha Srinivas of Northeastern Illinois University for her pioneering work on spin clusters and valuable comments on this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, X., Ito, H. & Torikai, E. A numerical study of spin-dependent organization of alkali-metal atomic clusters using density-functional method. J Nanopart Res 14, 1050 (2012). https://doi.org/10.1007/s11051-012-1050-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11051-012-1050-y