
Summary
Classical galactosaemia (McKusick 230400) is an: autosomal recessive disorder of galactose metabolism, caused by a deficiency of the enzyme galactose-1-phosphate uridyltransferase (GALT; EC 2.7.712). Most patients present in the neonatal period, after ingestion of galactose, with jaundice, hepatosplenomegaly, hepatocellular insufficiency, food intolerance, hypoglycaemia, renal tubular dysfunction, muscle hypotonia, sepsis and cataract. The gold standard for diagnosis of classical galactosaemia is measurement of GALT activity in erythrocytes. Gas-chromatographic determination of urinary sugars and sugar alcohols demonstrates elevated concentrations of galactose and galactitol. The only therapy for patients with classical galactosaemia is a galactose-restricted diet, and initially all galactose must be removed from the diet as soon as the diagnosis is suspected. After the neonatal period, a lactose-free diet is advised in most countries, without restriction of galactose-containing fruit and vegetables. In spite of the strict diet, long-term complications such as retarded mental development, verbal dyspraxia, motor abnormalities and hypergonadotrophic hypogonadism are frequently seen in patients with classical galactosaemia. It has been suggested that these complications may result from endogenous galactose synthesis or from abnormal galactosylation. Novel therapeutic strategies, aiming at the prevention of galactose 1-phosphate production, should be developed. In the meantime, the follow-up protocol for patients with GALT deficiency should focus on early detection, evaluation and, if possible, early intervention in problems of motor, speech and cognitive development.
Similar content being viewed by others

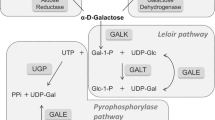

Abbreviations
- EGS:
-
endogenous galactose synthesis
- FSH:
-
follicle-stimulating hormone
- Gal-1-P:
-
galactose 1-phosphate
- GALE:
-
UDP-galactose epimerase
- GALK:
-
galactokinase
- GALM:
-
galactose mutarotase
- GALP:
-
galactose-1-phosphatase
- GALT:
-
galactose-1-phosphate uridyltransferase
- HRQoL:
-
Health Related Quality of Life
References
Acosta PB, Gross KC (1995) Hidden sources of galactose in the environment. Eur J Pediatr 154(Suppl 2): s87–92.
Ai Y, Zheng Z, O'Brien-Jenkins A, et al (2000) A mouse model of galactose-induced cataracts. Hum Mol Genet 9(12): 1821–1827.
Bandyopadhyay S, Chakrabarti J, Banerjee S, et al (2003) Prenatal exposure to high galactose adversely affects initial gonadal pool of germ cells in rats. Hum Reprod 18(2): 276–282.
Berry GT, Palmieri M, Gross KC, et al (1993) The effect of fruit and vegetables on urinary galactitol excretion in galactose-1-phosphate uridyltransferase deficiency. J Inherit Metab Dis 16: 91–100.
Berry GT, Nissim I, Lin Z, Mazur AT, Gibson JB, Segal S (1995) Endogenous synthesis of galactose in normal men and patients with hereditary galactosemia. Lancet 346: 1073–1074.
Berry GT, Leslie N, Reynolds R, Yager CT, Segal S (2001) Evidence for alternate galactose oxidation in a patient with deletion of the galactose-1-phosphate uridyltransferase gene. Mol Genet Metab 72(4): 316–321.
Berry GT, Moate PJ, Reynolds RA et al (2004) The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab 81: 22–30.
Boehles H, Wenzel D, Shin YS (1986) Progressive cerebellar and extrapyramidal motor disturbances in galactosaemic twins. Eur J Pediatrics 145: 413–417.
Bosch AM, Bakker HD, van Gennip AH, van Kempen JV, Wanders RJ, Wijburg FA (2002) Clinical features of galactokinase deficiency: a review of the literature. J Inherit Metab Dis 25(8): 629–634.
Bosch AM, Bakker HD, Maillette de Buy Wenniger-Prick LJ, Wanders RJA, Wijburg FA (2004a) High tolerance for oral galactose in classical galactosemia: dietary implications. Arch Dis Child 89(11): 1034–1036.
Bosch AM, Grootenhuis MA, Bakker HD, Heijmans HSA, Wijburg FA, Last BF (2004b) Living with classical galactosemia, health related quality of life consequences. Pediatrics 113(5): e423–e428.
Bosch AM, Ijlst L, Oostheim W, et al (2005) Identification of novel mutations in classical galactosemia. Hum Mutat 25(5): 502.
Cuatrecasas P, Segal S (1996) Galactose conversion to d-xylulose. An alternative route of galactose metabolism. Science 153(735): 549–551.
Elsas LJ, Dembure PP, Langley S, Paulk EM, Hjelm LN, Fridovich-Keil J (1994) A common mutation associated with the Duarte galactosemia allele. Am J Hum Genet 54: 1030–1036.
Elsas LJ, Lai K, Saunders CJ, Langley SD (2001) Functional analysis of the human galactose-1-phosphate uridyltransferase promoter in Duarte and LA variant galactosemia. Mol Genet Metab 72: 297–305.
Fox FP, Lucey JA, Cogan TM (1990) Glycolysis and related reactions during cheese manufacture and ripening. Crit Rev Food Sci Nutr 29(4): 237–253.
Gross KC, Acosta PB (1991) Fruit and vegetables are a source of galactose: implications of planning the diets of patients with galactosemia. J Inherit Metab Dis 14: 253–258.
Guerrero NV, Dingh RH, Manatunga A, Berry GT, Steiner RD, Elsas LJ (2000) Risk factors for premature ovarian failure in females with galactosemia. J Pediatr 137(6): 833–841.
Haberland C, Perou M, Brunngraber EG, Hof H (1971) The neuropathology of galactosemia. A histopathological and biochemical study. J Neuropathol Exp Neurol 30: 431–447.
Hirokawa H, Okano Y, Asada M, Fujimoto A, Suyama I, Isshiki G (1999) Molecular basis for phenotypic heterogeneity in galactosemia: prediction of clinical phenotype from genotype in Japanese patients. Eur J Hum Genet 7(7):757–764.
Holden HM, Rayment I, Thoden JB (2003) Structure and function of enzymes of the Leloir pathway for galactose metabolism. J Biol Chem 278(45): 43885–43888.
Holton JB (1995) Effects of galactosemia in utero. Eur J Pediatr 154(Supplement 2): s77–81.
Holton JB, Walter JH, Tyfield LA (2001) Galactosemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds; Childs B, Kinzler KW, Vogelstein B, assoc eds. The Metabolic and Molecular Basis of Inherited Disease, 8th edn. New York: McGraw-Hill, 1553–1583.
Honeyman MM, Green A, Holton JB, Leonard JV (1993) Galactosemia: results of the British Pediatric Surveillance Unit Study, 1988–90. Arch Dis Child 69: 339–341.
Huidekoper HH, Bosch AM, van der Crabben SN, Sauerwein HP, Ackermans MT, Wijburg FA (2005) Short-term exogenous galactose supplementation does not influence rate of appearance of galactose in patients with classical galactosemia. Mol Genet Metab 84(3): 265–272.
Hutchesson ACJ, Murdoch-Davies C, Green A, et al (1999) Biochemical monitoring of treatment for galactosemia: biological variability in metabolite concentrations. J Inherit Metab Dis 22: 138–148.
Huttenlocher PR, Hillman RE, Hsia YE (1970) Pseudotumor cerebri in classical galactosemia. J Pediatr 76(6): 902–905.
Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM (1956) Congenital galactosemia, a single enzymatic block in galactose metabolism. Science 123(3198): 635–636.
Isselbacher KJ (1957) Evidence for an accessory pathway of galactose metabolism in mammalian liver. Science 126: 652–654.
Jaeken J, Kint J, Spaapen LJM (1992) Serum lysosomal enzyme abnormalities in galactosemia. Lancet 340(8833): 1472–1473.
Jakobs C, Schweitzer S, Dorland B (1995) Galactitol in galactosemia. Eur J Pediatr 154 (Supplement 2): s50–52.
Kaufman FR, Kogut MD, Donnell GN, Goebelsmann U, March C, Koch R (1981) Hypergonadotropic hypogonadism in female patients with galactosemia. N Engl J Med 304: 994–998.
Kaufman FR, Loro ML, Azen C, Wenz E, Gilzans V (1993) Effect of hypogonadism and deficient calcium intake on bone density in patients with galactosemia. J Pediatr 123(3): 365–370.
Kaufman FR, McBride-Chang C, Manis FR, Wolff JA, Nelson MD (1995) Cognitive functioning, neurologic status and brain imaging in classic galactosemia. Eur J Pediatr 154(Supplement 2): s2–5.
Kobayashi RH, Kettelhut BV, Kobayashi AL (1983) Galactose inhibition of neonatal neutrophil function. Pediatr Infect Dis 2(6): 442–445.
Komrower GM, Lee DH (1970) Long term follow-up of galactosemia. Arch Dis Child 45: 367.
Lai K, Langley SD, Singh R, Dembuere PP, Hjelm LN, Elsas LJ (1996) A prevalent mutation for galactosemia among black Americans. J Pediatr 128: 89–95.
Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ (2003) GALT deficiency causes UDP hexose deficit in human galactosemic cells. Glycobiology 13(4): 285–294.
Langley SD, Lai K, Dembure PP, Hjelm LN, Elsas LJ (1997) Molecular basis for Duarte and Los Angeles variant galactosemia. Am J Hum Genet 60: 366–372.
Lebea PJ, Pretorius PJ (2005) The molecular relationship between deficient UDP-galactose uridyl transferase (GALT) and ceramide galactosyltransferase (CGT) enzyme function: a possible cause for poor long-term prognosis in classic galactosemia. Med Hypotheses 65(6): 1051–1057.
Lee PJ, Lilburn M, Wendel U, Schadewaldt P (2003) A woman with untreated galactosaemia. Lancet 362: 446.
Leslie N, Immerman E, Flach J, Florez M, Fridovich-Keil J, Elsas L (1992) The human galactose-1-phosphate uridyltransferase gene. Genomics 14: 474–480.
Levy HL, Driscoll SG, Porensky RS, Wender DF (1994) Ovarian failure in galactosemia. N Engl J Med 310: 50.
Lo W, Packmean S, Nash S, et al (1984) Curious neurologic sequelae in galactosemia. Pediatrics 73(3): 309–312.
Menezo YJR, Lescaille M, Nicollet B, Servy EJ (2004) Pregnancy and delivery after stimulation with rFSH of a galactosemia patient suffering hypergonadotropic hypogonadism: case report. J Assist Reprod Genet 3: 89–90.
Meyer WR, Doyle MB, Grifo JA, et al (1992) Aldose reductase inhibition prevents galactose-induced ovarian dysfunction in the Sprague-Dawley rat. Am J Obstet Gynecol 167: 1837–1843.
Nelson D (1995) Verbal dyspraxia in children with galactosemia. Eur J Pediatr 154 (suppl 2): s6–7.
Nelson MD, Wolff JA, Cross CA, Donnell GN, Kaufman FR (1982) Galactosemia: evaluation with MR imaging. Radiology 184: 255–261.
Ning C, Reynolds R, Chen J et al (2000) Galactose metabolism by the mouse with galactose-1-phosphate uridyltransferase deficiency. Pediatr Res 48(2): 211–217.
Novelli G, Reichardt JKV (2000) Molecular disorders of human galactose metabolism: past, present, and future. Mol Genet Metab 71: 662–665.
Ornstein KS, McGuire EJ, Berry GT, Roth S, Segal S (1992) Abnormal galactosylation of complex carbohydrates in cultured fibroblasts from patients with galactose-1-phosphate uridyltransferase deficiency. Pediatr Res 31(5): 508–511.
Panis B, Forget P, van Kroonenburgh MJ, et al (2004) Bone metabolism in galactosemia. Bone 35(4): 982–987.
Podskarbi T, Kohlmetz T, Gathof B, et al (1996) Molecular characterization of Duarte-1 and Duarte-2 variants of galactose-1-phosphate uridyltransferase. J Inherit Metab Dis 19: 638–644.
Prestoz LLC, Couto AS, Shin YS, Petry KG (1997) Altered follicle stimulated hormone isoforms in female galactosemia patients. Eur J Pediatr 156: 116–120.
Ridel KR, Leslie ND, Gilbert DL (2005) An updated 7 of the long term neurological effects of galactosemia. Pediatr Neurol 33(3): 153–161.
Robertson AMPH, Singh RH, Guerrero NV, Hundley M, Elsas LJ (2000) Outcome analysis of verbal dyspraxia in classic galactosemia. Genet Med 2(2): 142–148.
Rubio-Gozalbo ME, Hamming S, van Kroonenburg MJ, et al (2002) Bone mineral density in patients with classic galactosemia. Arch Dis Child 87(1): 57–60.
Schadewaldt P, Amalanathan L, Hammen HW, Wendel U (2004a) Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol Genet Metab 81: 31–44.
Schadewaldt P, Lilburn M, Wendel U, Lee P (2004b) Unexpected outcome in untreated galactosaemia. [Letter to the editor]. Mol Genet Metab 81: 255–257.
Schweitzer S, Shin Y, Jacobs C, Brodehl J (1993) Long-term outcome in 134 patients with galactosaemia. Eur J Paediatr 152: 36–43.
Schweitzer-Krantz S (2003) Early diagnosis of inherited metabolic disorders towards improving outcome: the controversial issue of galactosaemia. Eur J Pediatr 162(Supplement 1): S50–53.
Segal S (1998) Komrower Lecture. Galactosemia today: the enigma and the challenge. J Inherit Metab Dis 21(5): 455–471.
Segal S (2004) Another aspect of the galactosemic enigma. [Letter to the editor]. Mol Genet Metab 81: 253–254.
Shield JPH, Wadswordt EJK, MacDonald A, et al (2000) The relationship of genotype to cognitive outcome in galactosemia. Arch Dis Child 83: 248–250.
Shin-Buehring YS, Schaub J (1980) Pitfalls in the radioactive method of galactose-1-phosphate uridyltransferase activity measurement. Clin Chim Acta 106: 231–234.
Shin YS, Niedermeier HP, Endres W, Schaub J, Weidinger S (1987) Agarose gel isofocusing of UDP galactose pyrophosphorylase and galactose-1-phosphate uridyltrasferase. Developmental aspect of UDP-galactose pyrophosphorylase. Clin Chim Acta 166: 27–35.
Stambolian D (1988) Galactose and cataract. Surv Ophthalmol 32(5): 333–349.
Thompson SM, Netting MJ, Jerath S, Wiley V (2003) Effect of a less restricted diet in galactosemia. J Inherit Metab Dis 26(Supplement 2): 214.
Tyfield L, Carmichael D. The galactose-1-phosphate uridyltransferase mutation analysis database home page. (GALTdB) at http://www.ich.bris.ac.uk/galtdb/.
Tyfield L, Reichardt J, Fridovich-Keil J, et al (1999) Classical galactosemia and mutations at the galactose-1-phosphate uridyltransferase (GALT) gene. Hum Mutat 13: 417–430.
Vannas A, Hogan MJ, Golbus MS, Wood I (1975) Lens changes in a galactosemic fetus. Am J Ophthalmol 80(4): 726–733.
Waggoner DD, Buist NRM, Donnell GN (1990) Long-term prognosis in galactosemia: results of a survey of 350 cases. J Inherit Metab Dis 13: 802–818.
Webb AL, Singh RH, Kennedy MJ, Elsas LJ (2003) Verbal dyspraxia and galactosemia. Pediatr Res 53(3): 396–402.
Wehrli SL, Berry GT, Palmieri M, Mazur A, Elsas L, Segal S (1997) Urinary galactonate in patients with galactosemia: quantitation by nuclear magnetic resonance spectroscopy. Pediatr Res 42(6): 855–861.
Xu YK, Ng WG, Kaufman FR, Lobo RA, Donnell GN (1989) Galactose metabolism in human ovarian tissue. Pediatr Res 25(2): 151–155.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Georg Hoffmann
Competing interests: None declared
This article is based on a PhD thesis accepted by the University of Amsterdam, The Netherlands. Promotores: Prof. Dr. H. S. A. Heymans, Prof. Dr. R. J. Wanders. Co-promotores: Dr. H. D. Bakker, Prof. Dr. F. A. Wijburg
Electronic databases: Classical galactosaemia OMIM #230400 Galactose-1-phosphate uridyltransferase (GALT): EC 2.7.712
Rights and permissions
About this article
Cite this article
Bosch, A.M. Classical galactosaemia revisited. J Inherit Metab Dis 29, 516–525 (2006). https://doi.org/10.1007/s10545-006-0382-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-006-0382-0