
Abstract
The process of aging is complex involving numerous factors centered on transcriptional changes with advanced age. This study was aimed at elucidating mechanisms involved in mouse aging by conducting both gene expression and biochemical analyses on isolated mouse brain, heart and kidney. The gene expression analysis was not aimed at solely highlighting age-related transcriptional changes but also revealing regulated biological processes, cellular compartments, signaling and metabolic pathways. We have uncovered a conserved increase in the expression of genes mediating immune responses in all the tissues analyzed. In addition, elevated levels of lipid hydroperoxides (LPO)—an indicator of increased levels of radical oxygen species, implicate an oxidative stress-mediated activity of NF-kB signaling. In summary, these results suggest that transcriptional changes are most probably the downstream effect of environmental and endogenous factors constantly affecting the organism during its lifetime. In addition, we propose LPO as a potential biomarker of aging.
Similar content being viewed by others

Introduction
Research on the process of aging has revealed it’s complexity. This tenet is clearly demonstrated by the different theories of aging postulated over the last decades. Theories of aging fall into two broad categories: evolutionary models which pertain to the selective forces which determine species variation in life span, and proximate mechanisms, which determine the molecular process underlying individual variation in life span. Examples of the evolutionary models include the antagonistic pleiotropy theory introduced by Williams (1957) and the disposable soma theory proposed by Kirkwood (1977). Proximate mechanisms invoke a variety of molecular agents and include, for example, the free radical and mitochondrial decline theory (Harman 1956, 1972), the cross-linking theory (Sinex 1964), the Hayflick limit theory (Hayflick 1965), the membrane theory of aging (Zs-Nagy 1978), the neuroendocrine theory (Dilman et al. 1986), and the metabolic stability theory (Demetrius 2004). The implications of the free radical theory and the metabolic stability theory—models which involve ROS production rate and homeostasis, respectively—have been addressed using microarray-based analysis of gene expression. These empirical studies (Brink et al. 2008) have contributed to an increased awareness that aging is a systemic process which is driven by gene networks rather than a single-gene driven process (Budovskaya et al. 2008).
In mice, the first transcriptional profiles were generated in a limited number of mouse tissues such as muscle (Lee et al. 1999) and brain (Lee et al. 2000). Since then, transcriptional profiles of diverse organs and cells such as liver (Cao et al. 2001; Amador-Noguez et al. 2004; Fu et al. 2006), brain (Jiang et al. 2001; Fu et al. 2006), cardiomyocytes (Bahar et al. 2006), macrophages (Chelvarajan et al. 2006), heart (Dhahbi et al. 2006; Fu et al. 2006), adipose tissue (Higami et al. 2006), skeletal muscle (Edwards et al. 2007), and lungs (Misra et al. 2007) have been studied. In all of these studies, various tissues derived from mice of varying ages and strains were analyzed on varying platforms. A consequence of this is the noticeable differential expression in age-regulated target genes. A recent study (Zahn et al. 2007) now investigated and compared the transcriptional aging profiles of 16 mouse tissues. Overall, there is conservation of common biological processes related to aging, independent of tissue, strain, array platform etc. These processes include stress/immune responses, cell cycle, and metabolism.
In human, numerous studies have been conducted investigating aging in brain (Lu et al. 2004; Fraser et al. 2005), blood (Tan et al. 2005), eye (Segev et al. 2005), kidney (Rodwell et al. 2004; Melk et al. 2005), muscle (Welle et al. 2004; Giresi et al. 2005; Zahn et al. 2006), and skin (Lener et al. 2006). The available data suggests that age-related stress/immune responses, cell cycle, and metabolism are conserved.
In this work, we show by gene expression profiling of three mouse tissues (brain, heart and kidney) that activation of the immune response is a conserved age-related process. In addition, age-related increase in LPO concentrations may be accounted for by an oxidative stress-mediated activation of NF-kB signaling.
Materials and methods
Mice
Healthy wild type female C57BL6 mice were housed in a room with controlled photoperiod and temperature. Animals were given free access to water and pelleted diet. Mice were sacrificed by cervical dislocation; tissues were collected and flash frozen in liquid nitrogen and stored at −80°C. Brain, heart and kidneys were collected from young (8–10 weeks) and aged (17–19 month) mice. This study was approved by the institutional ethical committee.
RNA isolation
Whole mouse tissues (brain, heart and kidney) were homogenized in 1 ml Trizol (Invitrogen, Carlsbad, CA, USA) using the TissueLyser and 5 mm Stainless Steel Beads (Qiagen, Hilden, Germany) and homogenizing two times for 1 min at a frequency of 30.1/s. Homogenized samples were then incubated for 5 min at room temperature (RT). After addition of 200 μl chloroform, vortex mixing, incubation for 2 min at RT and centrifugation at 12,000g for 15 min at 4°C the water phase was transferred to a new tube. About 500 μl isopropanol were added and the mixture was incubated for 10 min at RT and afterwards centrifuged with 8,000g at 4°C for 5 min. The supernatant was discarded and the pellet washed with 1 ml of 75% ethanol, vortexed, and centrifuged at 8,000g for 5 min at 4°C. The supernatant was then discarded again and the pellet air dried for 10 min and afterwards re-suspended in 100 μl RNase-free water (dH2O). About 100 μg Trizol-extracted RNA in a volume of 100 μl was then purified with the RNeasy® Mini Kit (Qiagen). The procedure was performed following the manufacturers protocol including the also described DNase I on column treatment step to get rid of trace amounts of genomic DNA.
Illumina bead chip hybridisations and analysis of mouse expression data
Biotin-labeled cRNA was produced using a linear amplification kit (Ambion, Austin, TX, United States) with 400 ng of quality-checked total RNA as input. Chip hybridisations, washing, Cy3-streptavidin staining, and scanning were performed on the Illumina BeadStation 500 (Illumina, San Diego, CA, United States) platform using reagents and following protocols supplied by the manufacturer. cRNA samples were hybridised onto Illumina mouse-6 BeadChips. The following samples were hybridised as biological triplicates: young brain, aged brain, young heart, aged heart, young kidney and aged kidney. All basic expression data analysis was carried out using the manufacturer’s software BeadStudio 1.0. Raw data were background-subtracted and normalized using the “rank invariant” algorithm. Normalized data were then filtered for significant expression on the basis of negative control beads. Selection for differentially expressed genes was performed on the basis of arbitrary thresholds for fold changes plus statistical significance according to the Illumina t-test error model (Kuhn et al. 2004). Differentially expressed genes were further filtered according to Gene Ontology terms or mapped to Kegg pathways using DAVID 2006 (http://david.abcc.ncifcrf.gov). For analysis, we used GenBank accession numbers represented by the corresponding chip oligonucleotides as input. As this study is designed to find the potential involvement of pathways, biological processes and cellular compartments rather than to find aging marker genes, we used a ratio cut-off of 1.3-fold-change as it is of importance for the analysis of significant processes to provide a target gene list of hundreds of genes. In addition, overlapping target genes in all three tissues were analyzed using STRING (http://string.embl.de).
Real-time polymerase chain reaction (real-time PCR)
For reverse transcription using M-MLV reverse transcriptase (Promega, Madison, WI, USA), 2.0 μl (1 μg/μl) RNA was added to 0.5 μl of Oligo-dT primer (1 μg/μl; Invitek, Berlin, Germany) plus 7.5 μl of dH2O. The mixture was spun briefly, heated to 70°C for 3 min and cooled on ice. About 15.0 μl of master mix were added including the following components per reaction: 5.0 μl of 5× reaction buffer (Promega), 0.5 μl of (25 mM) dNTP, 0.1 μl of M-MLV reverse transcriptase (200 U/μl; Promega) and 9.4 μl of dH2O. After pulse spinning, incubation was carried out at 42°C for 1 h and then stopped at 65°C for 10 min. Real-time PCR was performed in 384-Well Optical Reaction Plates (Applied Biosystems, Foster City, CA, United States). The PCR mix in each well included 7.5 μl of SYBR®Green PCR Master Mix (Applied Biosystems), 0.25 μl dH2O, 2.25 μl of the forward and reverse primers (5 ng/μl; Invitek) and 5 μl of single strand cDNA (0.8 ng/μl) in a final reaction volume of 15 μl. Duplicate amplifications were carried out per gene with two wells serving as negative controls without template. Gapdh and Actb were amplified along with the target genes as endogenous controls for normalization. The PCR reaction was carried out on the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) using the following program, stage 1: 50°C for 2 min, stage 2: 95°C for 10 min, stage 3: 95°C for 15 s and 60°C for 1 min, for 40 cycles and, stage 4: 95°C for 15 s, 60°C for 15 s and 95°C for 15 s. The last heating step in stage 4 was performed with a ramp rate of 2% in order to enable the generation of a dissociation curve of the product. The output data generated by the Sequence Detection System 2 software were transferred to Excel (Microsoft, Redmond, WA, USA) for analysis. The differential mRNA expression of each gene was calculated with the comparative Ct (threshold cycle) method recommended by the manufacturer.
Western blot analysis
Tissue homogenates from the brain, heart and kidney (200 μg) of young and aged mice were separated by SDS–PAGE under reducing conditions and transferred onto nitrocellulose membranes. Membranes were blocked with 5% non fat dry milk for 1 h at room temperature. Antibodies used were ABCAM (Cambridge, UK) ab14232 (C3), ab16348 (donkey anti-chicken IgG) and Ambion (Darmstadt, Germany) #4300 (GAPDH). Membranes were then incubated overnight at 4°C with antibodies against C3 (1:20). The membranes were next incubated with peroxidase-labeled donkey anti-chicken IgG (1:4000) for 90 min, washed extensively and the protein bands visualized using ECL (ECL—Amersham, Buckinghamshire, UK) and exposure to ECL Hyperfilm.
Lipid hydroperoxide (LPO) measurement
Lipid hydroperoxides were extracted from duplicates of young and aged brain, heart and kidney using the Lipid hydroperoxide (LPO) Assay Kit (Calbiochem, Merck KGaA, Darmstadt, Germany)—following the manufacturers protocol. Whole tissues were homogenized in 500 μl dH2O using the TissueLyser and 5 mm Stainless Steel Beads (Qiagen) and homogenizing two times for 1 min and with a frequency of 30.1/s. After completion of the protocol the absorbance at 500 nm of each standard and sample was measured using the Ultrospec 3100 pro (GE Heathcare, Munich, Germany) and Chloroform:Methanol mixture as a blank. Results were calculated using a standard curve of the measured standards as indicated by the manufacturer.
Results
Global data analysis
We provide experimental data at the molecular level based on a genomics approach to investigate age-related transcriptional changes in mouse brain, heart and kidney. To identify and further analyze genes, pathways and biological processes altered with age in mouse, RNA was isolated from whole brain, heart and kidney of young (8–10 weeks) and aged (17–19 months) female mice. Whole-genome gene expression analysis employing the Illumina Bead Chip was used to profile the transcriptomes of these samples (full data available as Supplementary Table 1).
Data reproducibility is demonstrated by sample correlation and clustering (Fig. 1a, b). The three tissues show the expected separation from each other (R 2 < 0.50). However, the separation of the two age groups for each tissue is not explicit. In detail, replicates of young brain (R 2 = 0.98–0.99) and aged brain (R 2 = 0.97–1.00) show high correlations and also the correlation of young and aged samples is high (R 2 = 0.95–0.99). This high level of correlation makes it impossible to clearly separate the transcriptomes of a young brain to that from an aged brain.

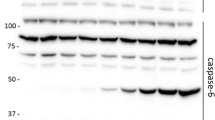
Results of the global expression data analysis. The figure shows a the sample clustering and b the corresponding correlation coefficients (R 2) derived from whole genome gene expression analyses for young and aged mouse whole brain, heart and kidney. Correlation coefficients are colored as follows: red = 0.90–1.00, orange = 0.80–0.90, white = 0.70–0.80 and gray <0.70. c Confirmation of microarray-derived data for relevant genes by Real-Time PCR. d Western blot analysis of complement component C3 on homogenates derived from brain, heart and kidney tissues (y: young; a: aged). In all three tissues we see an increased protein expression in the aged tissue compared to the respective young control. GAPDH was used as loading control (color figure in online)
Replicates of young heart (R 2 = 0.99–1.00) and aged heart (R 2 = 0.95–0.99) show better correlations than young compared to aged samples (R 2 = 0.84–0.95), however, one of the aged samples exhibited slight variations. Finally, replicates of young kidney (R 2 = 0.98–0.99) and aged kidney (R 2 = 0.96–0.98) show slightly better correlations than young compared to aged samples (R 2 = 0.94–0.98), leading to a clear separation of the three young from the two aged samples. The last aged sample exhibited a higher variation compared to the others. These differences could either reflect biological heterogeneity between samples or technical variations during the whole process of sample collection, RNA isolation and hybridisation.
Normalized data were analyzed for significant (detection >0.99 for at least one group and P-value <0.05) changes in gene expression between the two age groups with fold-changes of 1.3 and above (Supplementary Table 2). Using these criteria, we found regulated genes in all experiments with 828 regulated in brain, 990 in heart, and 1,544 in kidney. Activation or repression of gene expression was not observed in any of the tissues, as measured by a detection of more than 0.99 in the one age-group and a low detection of less than 0.8 in the other age-group. Real-Time PCR and Western blot analysis were employed to validate the changes in expression of relevant genes including the protein levels of C3 which is a component of the complement cascade. In accord with the C3 mRNA expression, the encoded protein also exhibited increased expression with age in brain heart and kidney (Fig. 1c, d).
Conservation of immune-related regulation in gene expression
As shown in Table 1, 828 genes were significantly regulated in mouse brain, 990 in heart and 1,544 in kidney. Of these, 581 exhibited increased and 247 decreased expression in brain, 569 increased and 421 decreased in heart and 1,112 genes showed increased and 432 decreased expression in kidney with age. In addition, DAVID pathway and Gene Ontology analyses revealed significantly regulated Kegg pathways and Gene Ontologies for up- and down-regulated genes in brain, heart and kidney (Supplementary Table 3). The conserved immune-related regulation in all three tissues will be highlighted in the ensuing paragraphs.
Table 2 lists genes exhibiting significant regulation with age in all three tissues with a minimum fold-ratio of 1.5. In total 28 genes were common in the target list of significant regulated genes in brain, heart and kidney. Of these, 25 genes showed increased and one exhibited decreased expression independent of tissue, whereas two genes showed distinct expression patterns. These two genes are Temt with increased expression in brain and heart and decreased expression in kidney and Tcrb-V8.2 with increased expression in heart and kidney and decreased expression in brain. STRING-analysis investigating the interactions of the overlapping target genes revealed a potential link to NF-kB (Fig. 2).

STRING analysis of the overlapping age-regulated target genes. The overlapping target genes from Table 2 were analyzed together with NF-kB (Rela) to show possible interactions. The figure shows that NF-kB is potentially connected to some of our conserved age-regulated target genes (e.g. Ii, Lyz, Ctss). The use of more interaction partners in the analysis also connects several other genes to the network (e.g. the complement components and the Fc-receptors; data not shown)
In addition to target gene comparisons, commonly regulated Kegg pathways and GOs were investigated by comparing the output lists. Table 3 lists Kegg pathways with significant regulation in at least 2 of the 3 tissues, and in addition biological processes and cellular compartments with similar regulation in all three tissues. Genes within five Kegg pathways relevant for immune responses exhibited increased expression in two or three tissues, whereas focal adhesion and glutathione metabolism showed different trends of regulation in heart and kidney. Several immune and inflammation-related GO-annotated biological processes showed increased expression.
Tissue-independent increase in oxidative stress with age
Based on the findings of our pathway analysis, which revealed the activation of the immune response, we decided to investigate the potential involvement of increased oxidative stress in aged tissues. A known effector of aging is the accumulation of ROS levels. With this in mind, we measured the level of ROS in our young and aged samples. Methods for measuring ROS levels directly are mainly established for cultured cells but not for tissues. Hence, we used a protocol for the measurement of LPO, which is a downstream product of ROS (Bartsch and Nair 2006). We could detect LPO in aged mouse tissues in contrast to young tissues where the levels were below the detection limit (Fig. 3). This implicates LPO as a potential biomarker of aging.

Lipid hydroperoxide (LPO) concentrations (μM) in extracts of young and aged mouse tissues. The figure shows that LPO could be detected in the aged brain, heart and kidney, but not in the corresponding young tissues. This result suggests a higher degree of oxidative stress in aged mice independent of the tissue. The concentrations of LPO were measured by the use of a standard curve method
Discussion
In this study, the process of aging in mouse tissues was investigated by analyzing age-associated transcriptional changes in brain, heart, and kidney. The analysis of the data shows that more genes exhibit age-related regulation in heart and kidney compared to the brain. A plausible explanation for this is that potentially a better protective system operates in brain as its function is of major importance for the organism. The detailed investigation of genes, biological processes, cellular compartments and Kegg-annotated pathways revealed an increase in immune-related processes in all three tissues. As this finding is very prominent and as this study is mainly focussed on uncovering conserved age-related processes, the discussion on mouse aging will be limited to overlapping findings in brain, heart, and kidney.
It was previously shown that aging is accompanied by a significant decline of adaptive immunity—immunosenescence (Hakim and Gress 2007), and an activation of innate immunity—inflammaging (Franceschi 2007). Interestingly, the age-related genes overlapping in the three tissues analyzed in this study all have functions related to the immune system and show in most cases increased expression with age. In addition, the overlap of biological processes and pathways confirms the increase in immune-related mechanisms. Recently, the activation of the innate immune system during aging was discussed to be a downstream effect of activated NF-kB signaling (Salminen et al. 2008) and the impact of NF-kB signaling on different mouse and human tissues was identified by an extensive screen for age-related transcription factor binding motifs (Adler et al. 2007). The validation of increased ROS levels with age in all three tissues observed in this study, the finding that ROS and oxidative stress can activate the NF-kB pathway (Schreck et al. 1992), and the interactions of NF-kB with a number of key target genes demonstrated by the STRING analysis (Fig. 2) implicates activated NF-kB signaling with aging. Increased expression of the genes relevant to the NF-kB complex was not observed in this study. However, the eventual activation of NF-kB signaling is underscored by the fact that activation of NF-kB is independent of expression changes of the NF-kB complex, but relies on its translocation from the cytoplasm to the nucleus in order to elicit activation or repression of target genes (Gosselin and Abbadie 2003). However, as female mice were investigated in this study, we cannot exclude the possibility of age-independent transcriptional changes resulting from cross-talk between estrogen receptor and NF-kB (Kalaitzidis and Gilmore 2005).
Nearly all the overlapping age-regulated genes in brain, heart and kidney exhibit increased expression with age. Because these genes are all related to the immune system, this leads to the speculation that they may play key roles in innate immunity. The innate immune system comprises the cells and mechanisms that defend the host from infection by other organisms in a non-specific manner. This means that the cells of the innate system recognize and respond to pathogens in a generic way. In contrast to the adaptive immune system, it does not confer long-lasting or protective immunity to the host. The major roles of the vertebrate innate immune system include the following.
Cytokine-mediated recruitment of immune cells
Inflammation is produced by eicosanoids and cytokines, which are released by injured or infected cells. Common cytokines include interleukins that are responsible for communication between white blood cells, chemokines that promote chemotaxis, and interferons that have anti-viral effects. In the list of overlapping age-related genes, at least five genes are present that are involved in the inflammatory response—Casp1, Irak3, Cd48, Dock2, and Icam1.
Casp1 was identified by its ability to proteolytically cleave and activate the inactive precursor of interleukin-1. The expression of its human homologue and the subsequent release of IL-1beta and IL-18 significantly contribute for instance to intestinal inflammation (Siegmund 2002). Moreover, it was recently concluded that IL-1beta and IL-18 participate in fundamental inflammatory processes that increase during aging (Dinarello 2006). Also directly related to IL-1 are the interleukin-1 receptor associate kinases, a member, Irak3, regulates innate immunity through unknown mechanism (Su et al. 2007). Cd48 is an interleukin (IL)-3-induced activating receptor on eosinophils, which may be involved in promoting allergic inflammation (Munitz et al. 2006). Dock2 is a member of chemokines that promote chemotaxis, which has been shown to be of key importance for lymphocyte chemotaxis (Fukui et al. 2001). Most interestingly, Icam1 is one of the proteins involved in inflammatory responses, it is overexpressed in senescent cells and aged tissues. Additionally, the NF-kB signaling cascade is crucial for the activation of human ICAM-1 expression in response to inflammation (Kletsas et al. 2004).
Activation of the complement cascade
The complement system is a biochemical cascade that attacks the surfaces of foreign cells. It contains over 20 different proteins and is named for its ability to “complement” the killing of pathogens by antibodies. Complement is the major humoral component of the innate immune response. This response is activated by complement binding to antibodies that have attached to microorganisms or the binding of complement proteins to carbohydrates on the surface of microorganisms. A group of genes from the list of overlapping age-related genes, known to be involved in the complement cascade are the five complement components—C1qa, C1qg, C3, C4, and Slp/C4a. Of these, the protein expression level of C3- a known target of NF-kB (Moon et al. 1999), increases with age (Fig. 1d). In addition, two genes encoding antibody proteins (Igh-1a and Igk-C) are present in the list, which may also contribute to this mechanism.
The detailed mechanisms of the complement cascade and the contribution of the single complement components is beyond the scope of this discussion. An important aspect of this work is the finding that several complement components exhibit differential gene expression with age (Plackett et al. 2004) and that these changes also contribute to age-related illnesses such as Alzheimer’s disease (Zhou et al. 2008).
Identification and removal of foreign substances by specialized white blood cells
Leukocytes (white blood cells) act like independent, single-cell organisms and are the cellular arm of the innate immune system. The innate leukocytes include the phagocytes (macrophages, neutrophils, and dendritic cells), mast cells, eosinophils, basophils, and natural killer cells. These cells identify and eliminate pathogens, either by attacking larger pathogens through contact or by engulfing and then killing microorganisms. The list of overlapping age-related genes includes at least three genes that are involved in phagocytosis—Fcgr3, Lyzs, and Cd68.
Phagocytosis is an important feature of cellular innate immunity performed by phagocytes that engulf pathogens. Fc-gamma receptors like Fcgr3 for instance, have major roles in the immune system such as phagocytosis (Nimmerjahn and Ravetch 2006). Lysozyme (Lyzs) serves as a non-specific innate opsonin by binding to the bacterial surface, and thus facilitating phagocytosis of the bacterium before opsonins from the acquired immune system arrive at the scene (Gomez and Balcazar 2008). Finally, macrosialin (Cd68) has been shown to have a specific function in macrophage-mediated phagocytosis (da Silva and Gordon 1999).
Activation of the adaptive immune system through antigen presentation
Antigen processing and presentation is 1 of the 3 overlapping pathways that is significantly activated with age in all three tissues. At least three genes are present in the overlapping list of age-related target genes involved in this process—Rmcs1/H2-Ab1, Ii/Cd74, and Ctss.
These genes play a role in major histocompatibility complex (MHC) class II antigen presentation. Rmcs1/H2-Ab1 is part of the MHC class II complex. The MHC class II-associated invariant chain (Ii) plays a critical role in the endocytic pathway, which is crucial in the MHC class II mediated antigen presentation (Rocha and Neefjes 2008). Furthermore, cathepsin S (Ctss) plays a major role in the degradation of the invariant peptide chain associated with the MHC and thus also affecting antigen presentation (Gupta et al. 2008). One should bear in mind that the above-mentioned genes are not restricted to their described functions.
Furthermore, as the innate and adaptive immune systems have overlapping functions it is also likely that the genes involved have several roles in immunity. An increase in the adaptive immune system for instance may contribute to autoimmunity. For example, a case-control study of human subjects suffering from rheumatoid arthritis identified an associated single nucleotide polymorphism in the NCF4 gene, supporting a role in autoimmunity (Olsson et al. 2007). Ncf4 is among the conserved age-related genes of this study. Additionally, certain autoimmune responses are known to increase with age (Yung and Julius 2008).
In summary, the analysis of transcriptional profiles of aging in the three mouse tissues, brain, heart, and kidney, reveals activation of the immune response as a conserved mechanism in mouse aging. We propose the following: (a) LPO as a potential biomarker of aging (Fig. 3) and (b) a scheme whereby increased levels of ROS activate the immune response via NF-kB signaling (Fig. 4).

ROS-mediated activation of innate immunity. The figure presents a model of ROS-mediated activation of innate immunity via the NF-kB signaling pathway. As the activation of innate immunity was shown by transcriptional analysis and the increase of oxidative stress by the measurement of LPO, the involvement of NF-kB is hypothetical and based on literature (Schreck et al. 1992; Gosselin and Abbadie 2003; Salminen et al. 2008)
ROS thus plays a critical role in the aging process. The free radical theory and the metabolic stability are two competing models which propose proximate mechanisms of the dynamics of aging. Doonan et al. (2008) has used the model organism C. elegans to provide additional critique of the free radical theory. The work of Doonan et al. (2008), when integrated with the studies in this article and the analysis given in Brink et al. (2008) are consistent with the proposition that maintaining homeostatic levels of ROS determine the rate of aging. These studies furthermore establish that aging does not depend on the activity of a single gene but on the aggregated action of several gene networks.
References
Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY (2007) Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev 21:3244–3257. doi:10.1101/gad.1588507
Amador-Noguez D, Yagi K, Venable S, Darlington G (2004) Gene expression profile of long-lived Ames dwarf mice and little mice. Aging Cell 3:423–441. doi:10.1111/j.1474-9728.2004.00125.x
Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J (2006) Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441:1011–1014. doi:10.1038/nature04844
Bartsch H, Nair J (2006) Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg 391:499–510. doi:10.1007/s00423-006-0073-1
Brink TC, Demetrius L, Lehrach H, Adjaye J (2008) Age-related transcriptional changes in gene expression in different organs of mice support the metabolic stability theory of aging. Biogerontology. doi:10.1007/s10552-008-9197-8
Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK (2008) An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell 134:291–303. doi:10.1016/j.cell.2008.05.044
Cao SX, Dhahbi JM, Mote PL, Spindler SR (2001) Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc Natl Acad Sci USA 98:10630–10635. doi:10.1073/pnas.191313598
Chelvarajan RL, Liu Y, Popa D, Getchell ML, Getchell TV, Stromberg AJ, Bondada S (2006) Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leukoc Biol 79:1314–1327. doi:10.1189/jlb.0106024
da Silva RP, Gordon S (1999) Phagocytosis stimulates alternative glycosylation of macrosialin (mouse CD68), a macrophage-specific endosomal protein. Biochem J 338(Pt 3):687–694. doi:10.1042/0264-6021:3380687
Demetrius L (2004) Caloric restriction, metabolic rate, and entropy. J Gerontol A Biol Sci Med Sci 59:B902–B915
Dhahbi JM, Tsuchiya T, Kim HJ, Mote PL, Spindler SR (2006) Gene expression and physiologic responses of the heart to the initiation and withdrawal of caloric restriction. J Gerontol A Biol Sci Med Sci 61:218–231
Dilman VM, Revskoy SY, Golubev AG (1986) Neuroendocrine-ontogenetic mechanism of aging: toward an integrated theory of aging. Int Rev Neurobiol 28:89–156. doi:10.1016/S0074-7742(08)60107-5
Dinarello CA (2006) Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr 83:447S–455S
Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, Matscheski A, Vanfleteren JR, Gems D (2008) Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev 22(23):3236–3241. doi:10.1101/gad.504808
Edwards MG, Anderson RM, Yuan M, Kendziorski CM, Weindruch R, Prolla TA (2007) Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics 8:80. doi:10.1186/1471-2164-8-80
Franceschi C (2007) Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr Rev 65:S173–S176. doi:10.1111/j.1753-4887.2007.tb00358.x
Fraser HB, Khaitovich P, Plotkin JB, Paabo S, Eisen MB (2005) Aging and gene expression in the primate brain. PLoS Biol 3:e274. doi:10.1371/journal.pbio.0030274
Fu C, Hickey M, Morrison M, McCarter R, Han ES (2006) Tissue specific and non-specific changes in gene expression by aging and by early stage CR. Mech Ageing Dev 127:905–916. doi:10.1016/j.mad.2006.09.006
Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, Inayoshi A, Noda M, Oike M, Shirai T, Sasazuki T (2001) Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature 412:826–831. doi:10.1038/35090591
Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, Kandarian SC (2005) Identification of a molecular signature of sarcopenia. Physiol Genomics 21:253–263. doi:10.1152/physiolgenomics.00249.2004
Gomez GD, Balcazar JL (2008) A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol Med Microbiol 52:145–154. doi:10.1111/j.1574-695X.2007.00343.x
Gosselin K, Abbadie C (2003) Involvement of Rel/NF-kappa B transcription factors in senescence. Exp Gerontol 38:1271–1283. doi:10.1016/j.exger.2003.09.007
Gupta S, Singh RK, Dastidar S, Ray A (2008) Cysteine cathepsin S as an immunomodulatory target: present and future trends. Expert Opin Ther Targets 12:291–299. doi:10.1517/14728222.12.3.291
Hakim FT, Gress RE (2007) Immunosenescence: deficits in adaptive immunity in the elderly. Tissue Antigens 70:179–189. doi:10.1111/j.1399-0039.2007.00891.x
Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11:298–300
Harman D (1972) The biologic clock: the mitochondria? J Am Geriatr Soc 20:145–147
Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37:614–636. doi:10.1016/0014-4827(65)90211-9
Higami Y, Barger JL, Page GP, Allison DB, Smith SR, Prolla TA, Weindruch R (2006) Energy restriction lowers the expression of genes linked to inflammation, the cytoskeleton, the extracellular matrix, and angiogenesis in mouse adipose tissue. J Nutr 136:343–352
Jiang CH, Tsien JZ, Schultz PG, Hu Y (2001) The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc Natl Acad Sci USA 98:1930–1934. doi:10.1073/pnas.98.4.1930
Kalaitzidis D, Gilmore TD (2005) Transcription factor cross-talk: the estrogen receptor and NF-kappaB. Trends Endocrinol Metab 16:46–52. doi:10.1016/j.tem.2005.01.004
Kirkwood TB (1977) Evolution of ageing. Nature 270:301–304. doi:10.1038/270301a0
Kletsas D, Pratsinis H, Mariatos G, Zacharatos P, Gorgoulis VG (2004) The proinflammatory phenotype of senescent cells: the p53-mediated ICAM-1 expression. Ann N Y Acad Sci 1019:330–332. doi:10.1196/annals.1297.056
Kuhn K, Baker SC, Chudin E, Lieu MH, Oeser S, Bennett H, Rigault P, Barker D, McDaniel TK, Chee MS (2004) A novel, high-performance random array platform for quantitative gene expression profiling. Genome Res 14:2347–2356. doi:10.1101/gr.2739104
Lee CK, Klopp RG, Weindruch R, Prolla TA (1999) Gene expression profile of aging and its retardation by caloric restriction. Science 285:1390–1393. doi:10.1126/science.285.5432.1390
Lee CK, Weindruch R, Prolla TA (2000) Gene-expression profile of the ageing brain in mice. Nat Genet 25:294–297. doi:10.1038/77046
Lener T, Moll PR, Rinnerthaler M, Bauer J, Aberger F, Richter K (2006) Expression profiling of aging in the human skin. Exp Gerontol 41:387–397. doi:10.1016/j.exger.2006.01.012
Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429:883–891. doi:10.1038/nature02661
Melk A, Mansfield ES, Hsieh SC, Hernandez-Boussard T, Grimm P, Rayner DC, Halloran PF, Sarwal MM (2005) Transcriptional analysis of the molecular basis of human kidney aging using cDNA microarray profiling. Kidney Int 68:2667–2679. doi:10.1111/j.1523-1755.2005.00738.x
Misra V, Lee H, Singh A, Huang K, Thimmulappa RK, Mitzner W, Biswal S, Tankersley CG (2007) Global expression profiles from C57BL/6J and DBA/2J mouse lungs to determine aging-related genes. Physiol Genomics 31:429–440
Moon RM, Parikh AA, Pritts TA, Fischer JE, Cottongim S, Szabo C, Salzman AL, Hasselgren P-O (1999) Complement component C3 production in IL-1beta-stimulated human intestinal epithelial cells is blocked by NF-kappaB inhibitors and by transfection with ser 32/36 mutant IkappaBalpha. J Surg Res 82:48–55. doi:10.1006/jsre.1998.5503
Munitz A, Bachelet I, Eliashar R, Khodoun M, Finkelman FD, Rothenberg ME, Levi-Schaffer F (2006) CD48 is an allergen and IL-3-induced activation molecule on eosinophils. J Immunol 177:77–83
Nimmerjahn F, Ravetch JV (2006) Fcgamma receptors: old friends and new family members. Immunity 24:19–28. doi:10.1016/j.immuni.2005.11.010
Olsson LM, Lindqvist AK, Kallberg H, Padyukov L, Burkhardt H, Alfredsson L, Klareskog L, Holmdahl R (2007) A case-control study of rheumatoid arthritis identifies an associated single nucleotide polymorphism in the NCF4 gene, supporting a role for the NADPH-oxidase complex in autoimmunity. Arthritis Res Ther 9:R98. doi:10.1186/ar2299
Plackett TP, Boehmer ED, Faunce DE, Kovacs EJ (2004) Aging and innate immune cells. J Leukoc Biol 76:291–299. doi:10.1189/jlb.1103592
Rocha N, Neefjes J (2008) MHC class II molecules on the move for successful antigen presentation. EMBO J 27:1–5. doi:10.1038/sj.emboj.7601945
Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, Myers BD, Brooks JD, Davis RW, Higgins J, Owen AB, Kim SK (2004) A transcriptional profile of aging in the human kidney. PLoS Biol 2:e427. doi:10.1371/journal.pbio.0020427
Salminen A, Huuskonen J, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2008) Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev 7:83–105. doi:10.1016/j.arr.2007.09.002
Schreck R, Albermann K, Baeuerle PA (1992) Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic Res Commun 17:221–237. doi:10.3109/10715769209079515
Segev F, Mor O, Segev A, Belkin M, Assia EI (2005) Downregulation of gene expression in the ageing lens: a possible contributory factor in senile cataract. Eye 19:80–85. doi:10.1038/sj.eye.6701423
Siegmund B (2002) Interleukin-1beta converting enzyme (caspase-1) in intestinal inflammation. Biochem Pharmacol 64:1–8. doi:10.1016/S0006-2952(02)01064-X
Sinex FM (1964) Cross-linkage and aging. Adv Gerontol Res 21:165–180
Su J, Xie Q, Wilson I, Li L (2007) Differential regulation and role of interleukin-1 receptor associated kinase-M in innate immunity signaling. Cell Signal 19:1596–1601. doi:10.1016/j.cellsig.2007.02.009
Tan Q, Christensen K, Christiansen L, Frederiksen H, Bathum L, Dahlgaard J, Kruse TA (2005) Genetic dissection of gene expression observed in whole blood samples of elderly Danish twins. Hum Genet 117:267–274. doi:10.1007/s00439-005-1308-x
Welle S, Brooks AI, Delehanty JM, Needler N, Bhatt K, Shah B, Thornton CA (2004) Skeletal muscle gene expression profiles in 20–29 year old and 65–71 year old women. Exp Gerontol 39:369–377. doi:10.1016/j.exger.2003.11.011
Williams GC (1957) Pleiotropy, natural selection and the evolution of senescence. Evol Int J Org Evol 11:398–411. doi:10.2307/2406060
Yung RL, Julius A (2008) Epigenetics, aging, and autoimmunity. Autoimmunity 41:329–335. doi:10.1080/08916930802024889
Zahn JM, Sonu R, Vogel H, Crane E, Mazan-Mamczarz K, Rabkin R, Davis RW, Becker KG, Owen AB, Kim SK (2006) Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet 2:e115. doi:10.1371/journal.pgen.0020115
Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, Lakatta EG, Boheler KR, Xu X, Mattson MP, Falco G, Ko MS, Schlessinger D, Firman J, Kummerfeld SK, Wood WH, Zonderman AB, Kim SK, Becker KG (2007) AGEMAP: a gene expression database for aging in mice. PLoS Genet 3:e201. doi:10.1371/journal.pgen.0030201
Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ (2008) Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s Disease. J Neurochem 106:2080–2092
Zs-Nagy I (1978) A membrane hypothesis of aging. J Theor Biol 75:189–195. doi:10.1016/0022-5193(78)90230-8
Acknowledgments
This study was supported by the Max Planck Society, the Federal Ministry of Education and Research (BMBF) as part of the German National Genome Research Network (NGFN-II/EP0313359) program and the LEHRACH-STIFTUNG zur Förderung der Genomforschung.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Brink, T.C., Regenbrecht, C., Demetrius, L. et al. Activation of the immune response is a key feature of aging in mice. Biogerontology 10, 721–734 (2009). https://doi.org/10.1007/s10522-009-9219-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-009-9219-1