
Abstract
We present a quasi-continuum self-diffusion theory that can capture the ordering effects and the density variations that are predicted by non-equilibrium molecular dynamics (NEMD) in nanochannel flows. A number of properties that affect fluid ordering in NEMD simulations are extracted and compared with the quasi-continuum predictions. The proposed diffusion equation requires the classic diffusion coefficient D and a micro structural internal length g that relates directly to the shape of the molecular potential of the NEMD calculations. The quasi-continuum self-diffusion theory comes as an alternative to atomistic simulation, bridging the gap between continuum and atomistic behavior with classical hydrodynamic relations and reduces the computational burden as compared with fully atomistic simulations.





Similar content being viewed by others

Notes
If a gravitational field \(\phi\) is present, we replace \(\mu\) with \(\mu + m\phi\) (\(m\) is the mass per mole). For \(\phi = {\text{const}}\), the present results will still hold true.
Abbreviations
- A 1–4 :
-
Constants determined by BC
- A, B :
-
Constants for inhomogeneous diffusion
- A i , B i :
-
Airy functions
- c :
-
Concentration
- c 1–3 :
-
Real constants
- D :
-
Bulk diffusion coefficient
- D ap :
-
Apparent diffusion coefficient
- F :
-
Diffusion functional
- F ap :
-
Apparent diffusion functional
- 1 F 1 :
-
Hypergeometric function
- F ext :
-
Magnitude of external driving force
- g :
-
Wavelength
- G :
-
Gibbs free energy
- G 0 :
-
Gibbs free energy at equilibrium
- h :
-
Boundary value for concentration
- H:
-
Hermitian polyomial
- h ch :
-
Channel height
- J :
-
Diffusional flux
- K :
-
Gradient energy coefficient
- K * :
-
Spring constant
- L x :
-
Length of the computational domain in the x-direction
- L y :
-
Length of the computational domain in the y-direction
- L z :
-
Length of the computational domain in the z-direction
- M :
-
Diffusional mobility
- n :
-
Integer number, n = 0, 1, 2…
- p :
-
Pressure
- q :
-
Boundary value for the non-classic flux term
- r eq :
-
Position of a wall atom on fcc lattice site
- r i :
-
Position vector of atom i
- r ij :
-
Distance vector between ith and jth atom
- T :
-
Temperature
- S :
-
Area
- u(r ij ):
-
LJ potential of atom i with atom j
- V :
-
Volume
- w :
-
Boundary condition for the normal flux
- z * :
-
Normalized distances in the z-direction
- ε :
-
Energy parameter in the LJ potential
- μ :
-
Local chemical potential
- ρ :
-
Fluid density
- σ :
-
Length parameter in the LJ potential
References
Allen MP, Tildesley TJ (1987) Computer simulation of liquids. Clarendon Press, Oxford
Asproulis N, Drikakis D (2011) Wall-mass effects on hydrodynamic boundary slip. Phys Rev E 84:031504
Binder K, Horbach J, Kob W, Paul W, Varnik F (2004) Molecular dynamics simulations. J Phys: Condens Matter 16:429–453
Cahn JW (1961) On spinodal decomposition. Act Meta 9:765–801
Cahn JW, Hilliard JE (1958) Free energy of a non uniform system. I. Interfacial free energy. J Chem Phys 28:258–267
Cahn JW, Hilliard JE (1959) Free energy of a non uniform system. III. Nucleation in a two-component incompressible fluid. J Chem Phys 31:688–699
Cook HE (1970) Brownian motion in spinodal decomposition. Act Meta 18:297–306
Courant R, Hilbert D (1953) Methods of mathematical physics, vol I. Wiley Interscience, New York
Crank J (1975) The mathematics of diffusion. Oxford University Press, New York
Darken LS (1948) Diffusion, mobility and their interrelation through free energy in binary metallic systems. Trans AIME 175:184–201
Evans DJ, Holian BL (1985) The Nosé–Hoover thermostat. J Chem Phys 83:4069–4074
Fouladi F, Hosseinzadeh E, Barari A, Dmairry G (2010) Highly nonlinear temperature-dependent fin: analysis by variational iteration method. Heat Tranfer Res 41:1–11
Gini-Castagnoli G, Ricci FP (1960) Self-diffusion in liquid argon. J Chem Phys 32:19–20
Hartkamp R, Ghosh A, Weinhart T, Luding S (2012) A study of the anisotropy of stress in a fluid confined in a nanochannel. J Chem Phys 137:044711
Holian BL, Voter AF (1995) Thermostatted molecular dynamics: How to avoid the Toda demon hidden in Nosé–Hoover dynamics. Phys Rev E 52:2338–2347
Iwai Y, Uchida H, Koga Y, Arai Y, Movi Y (1996) Monte Carlo simulation of solubilities of aromatic compounds in supercritical carbon dioxide by a group contribution site model. Ind Eng Chem Res 35:3782–3787
Li Q, Liu C (2012) Molecular dynamics simulation of heat transfer with effects of fluid–lattice interactions. Int J Heat Mass Trans 55:8088–8092
Lipowsky R, Fisher ME (1986) Wetting in random systems. Phys Rev Lett 56:472–475
Lipowsky R, Huse DA (1986) Diffusion-limited growth of wetting layers. Phys Rev Lett 57:353–356
Marko JF (1993) Influence of surface interactions on spinodal decomposition. Phys Rev E 48:2861–2879
Nagayama G, Cheng P (2004) Effects of interface wettability on microscale flow by molecular dynamics simulation. Int J Heat Mass Trans 47:501–513
Priezjev NV (2007) Effect of surface roughness on rate-dependent slip in simple fluids. J Chem Phys 127:144708
Prigogine I, Glansdorff P (1965) Variational properties and fluctuation theory. Physica 31:1242–1256
Sofos F, Karakasidis TE, Liakopoulos A (2009a) Non-equilibrium molecular dynamics investigation of parameters affecting planar nanochannel flows. Cont Eng Sci 2:283–298
Sofos F, Karakasidis TE, Liakopoulos A (2009b) Transport properties of liquid argon in krypton nanochannels: anisotropy and non-homogeneity introduced by the solid walls. Int J Heat Mass Trans 52:735–743
Sofos F, Karakasidis TE, Liakopoulos A (2012) Surface wettability effects on flow in rough wall nanochannels. Microfluid Nanofluid 12:25–31
Somers SA, Davis HT (1991) Microscopic dynamics of fluids confined between smooth and atomically structured solid surfaces. J Chem Phys 96:5389–5407
Stanley HE (1971) Introduction to phase transitions and critical phenomena. Clarendon, Oxford
Thomas JA, McGaughey AJH (2007) Effect of surface wettability on liquid density, structure, and diffusion near a solid surface. J Chem Phys 126:034707
Voronov RS, Papavassiliou DV, Lee LL (2006) Boundary slip and wetting properties of interfaces: correlation of the contact angle with the slip length. J Chem Phys 124:204701
Acknowledgments
This project was implemented under the “ARISTEIA II” Action of the “OPERATIONAL PROGRAMME EDUCATION AND LIFELONG LEARNING” and is co-founded by the European Social Fund (ESF) and National Resources.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix 1: ODE solution for the 1D state problem
The general solution of Eq. (30) is
where A 1, …A 4 are constants to be determined by BC. The first two terms are similar to the classic solution. The other two terms are the non-classic part of the solution. Taking the region 0 ≤ z ≤ h ch, (h ch is the channel width), we impose the following BCs that are compatible with the Poiseuille flow in the normal to z-direction.
At z = 0 and h ch, J = 0, that is no flux at the walls, which gives
At z = h ch/2, c = c o a datum point at the middle of the channel (can be thought of as a constant that increases with temperature), which gives
Symmetry of the concentration implies dc/dz = 0 at z = h ch/2, which gives
This condition implies that J = 0 at z = h ch, as expected. It can be shown that J = 0 for all z, 0 ≤ z ≤ h ch.
Mass conservation implies
which gives
Solving Eqs. (49), (50) and (52), we obtain
For \(\bar{c} = c_{o}\), we obtain the classic relation c = c o .
Appendix 2: Asymptotic analysis
The solution of Eq. (37) can be written as combinations of hypergeometric function 1 F 1 and the Hermitian polynomial H:
where c 1 and c 2 are real constants. Note that only the hypergeometric function 1 F 1 is symmetric with respect to z. Therefore, symmetric cases are accepted only if c 1 = 0. An interesting expansion of the solution around z = 0 (middle of the channel) is given below. In some cases in the field of heat transfer, the variational iteration method is utilized as an approximate analytical method to overcome some inherent limitations arising as uncontrollability to the nonzero endpoint boundary conditions (see, for example, Fouladi et al. 2010).
It is of interest to expand the symmetric solution (c 1 = 0) around point z = 0 (the middle of the channel width).
Note that the expansion (57) involves only even powers of z. Taking the conditions dc/dz = 0 and c = c o at z = 0, and retaining the first two terms of (57), we obtain
and
Assuming that (59) should provide c ≥ 0 for all values of c o ≥ 0, we conclude that c 2 ≥ 0. In other words, dc 2/dz 2 ≥ 0 at z = 0, a result that is verified by the NEMD calculations!
Appendix 3: Diffusion for asymmetric wall conditions
The case of asymmetric wall conditions can be studied by a diffusion inhomogeneous distribution of the type
The diffusion equation becomes
The solution of Eq. (61) can be put in closed form.
where A i and B i are Airy functions and c 2, c 3 are constants. The full solution of Eq. (62) is given by
where c o , c 1, c 2, c 3 are constants.
The general form of the functions C A(z) and C B(z)are
p F q is the generalized hypergeometric function, I is the modified Bessel function of the first kind, Γ is the Gamma function, and B is a parameter of the problem.
We believe that the chemical affinity between the walls and the fluid, as well as the roughness of the walls, influence the diffusion coefficient distribution and this, in turn, affects the steady state concentration profiles.
Appendix 4: Temperature profile
Temperature is calculated in each bin across the channel using
where N bin is the number of fluid atoms in the bin examined, i = 1, 2, 3, … denotes the x, y, z component of the atomic velocity \(v_{n - 1}\), \({\bar{\text{v}}}_{\text{i}}\) is the ith component of the mean macroflow velocity, k B is the Boltzman constant, and m Ar is argon atom mass.
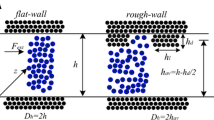
In Fig. 6a, the calculated fluid temperature distribution for h ch = 6.3 nm is presented. We can see that for each system temperature (the temperature value imposed at the wall’s thermostats, 100, 120, or 150 K), the temperature profile is approximately flat at this temperature value. We obtain similar flat behavior when we vary system parameters such as channel width h ch, the wall atoms spring constant K *, the wall–fluid interaction ratio ɛ wall /ɛ fluid an the average fluid density ρ *. However, in Fig. 6b, for h ch = 17.1 nm and external forces above 0.036 pN, temperature profiles are not uniform and show a rise in the middle of the channel and near the solid walls. Similar behavior is reported on Nagayama and Cheng (2004) when the external force that drives the flow becomes large. We attribute this behavior to the fact that at large channel widths, in combination with external forces of large magnitude, strain rates inside the nanochannel increase, and the system has non-linear response.

Temperature profiles a for system temperature T = 100, 120 and 150 K, and h ch = 6.3 nm b for various magnitudes of the F ext (normalized), T = 120 K and \(h_{\text{ch}} = 17.1{\text{ nm}}\)
Rights and permissions
About this article
Cite this article
Giannakopoulos, A.E., Sofos, F., Karakasidis, T.E. et al. A quasi-continuum multi-scale theory for self-diffusion and fluid ordering in nanochannel flows. Microfluid Nanofluid 17, 1011–1023 (2014). https://doi.org/10.1007/s10404-014-1390-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10404-014-1390-2