
Abstract
Immunosuppression plays a pivotal role in assisting tumors to evade immune destruction and promoting tumor development. We hypothesized that genetic variation in the immunosuppression pathway genes may be implicated in breast cancer tumorigenesis. We included 42,510 female breast cancer cases and 40,577 controls of European ancestry from 37 studies in the Breast Cancer Association Consortium (2015) with available genotype data for 3595 single nucleotide polymorphisms (SNPs) in 133 candidate genes. Associations between genotyped SNPs and overall breast cancer risk, and secondarily according to estrogen receptor (ER) status, were assessed using multiple logistic regression models. Gene-level associations were assessed based on principal component analysis. Gene expression analyses were conducted using RNA sequencing level 3 data from The Cancer Genome Atlas for 989 breast tumor samples and 113 matched normal tissue samples. SNP rs1905339 (A>G) in the STAT3 region was associated with an increased breast cancer risk (per allele odds ratio 1.05, 95 % confidence interval 1.03–1.08; p value = 1.4 × 10−6). The association did not differ significantly by ER status. On the gene level, in addition to TGFBR2 and CCND1, IL5 and GM-CSF showed the strongest associations with overall breast cancer risk (p value = 1.0 × 10−3 and 7.0 × 10−3, respectively). Furthermore, STAT3 and IL5 but not GM-CSF were differentially expressed between breast tumor tissue and normal tissue (p value = 2.5 × 10−3, 4.5 × 10−4 and 0.63, respectively). Our data provide evidence that the immunosuppression pathway genes STAT3, IL5, and GM-CSF may be novel susceptibility loci for breast cancer in women of European ancestry.
Similar content being viewed by others

Introduction
Breast cancer is the most frequent cancer among women and the second leading cause of cancer-related death after lung cancer in Europe. In addition to genetic variants with high and moderate penetrance, more than 90 common germline genetic variants contributing to breast cancer risk have been identified, comprising about 37 % of the familial relative risk of the disease (Michailidou et al. 2013, 2015). This suggests that a substantial portion of inherited variation has not yet been identified. In addition, most of the known common susceptibility variants reside in non-coding regions and result in subtle regulation of gene expression. The biological mechanisms through which genetic variants exert their functions are still not entirely understood.
The ability to evade immune destruction has been increasingly recognized as a key hallmark of tumors (Hanahan and Weinberg 2011). Tumor cells may secrete immunosuppressive factors like TGF-β which hampers infiltrating cytotoxic T lymphocytes and natural killer cells (Yang et al. 2010). Inflammatory cells like regulatory T cells (Treg cells), a subset of CD4+ T lymphocytes, as well as myeloid-derived suppressor cells (MDSCs) may be recruited into the tumor environment, which are actively immunosuppressive (Lindau et al. 2013; Reisfeld 2013). Higher prevalence of Treg cells has been found in various cancers (Chang et al. 2010; Michel et al. 2008; Watanabe et al. 2002), including breast cancer (Bates et al. 2006). There is evidence that tumor infiltrating Treg cells endowed with immunosuppressive potential are associated with tumor progression and unfavorable prognosis, especially in estrogen receptor (ER)-negative breast cancer (Bates et al. 2006; Kim et al. 2013; Liu et al. 2012a). In addition, infiltrating MDSCs were also found in murine mammary tumor models (Aliper et al. 2014; Gad et al. 2014), but their relevance for breast cancer patients also in terms of prognosis is not well-understood. Furthermore, previous association studies have identified susceptibility alleles for breast cancer in two genes, TGFBR2 (transforming growth factor beta receptor II) (Michailidou et al. 2013) and CCND1 (cyclin D1) (French et al. 2013), which may be involved in immune regulation in cancer patients (Gabrilovich and Nagaraj 2009; Krieg and Boyman 2009), including those with breast cancer. We hypothesized that immunosuppression pathway genes, particularly those relevant to Treg cell and MDSC functions, may harbor further susceptibility variants associated with breast cancer tumorigenesis, with a possible differential association by ER status.
In this analysis, we investigated associations between breast cancer risk and single nucleotide polymorphisms (SNPs) in 133 candidate genes in the immunosuppression pathway in individual level data from the Breast Cancer Association Consortium (BCAC). We also assessed associations with breast cancer risk at the gene and pathway levels. Furthermore, we used publicly available datasets through the UCSC Genome Browser (2015) to examine the putative genetic susceptibility loci for potential regulatory function.
Materials and methods
Study participants
In this analysis, participants were restricted to 83,087 women of European ancestry from 37 case–control studies participating in BCAC, including 42,510 invasive breast cancer cases with stage I–III disease and 40,577 cancer-free controls. Of all breast cancer patients, 26,094 were known to have ER-positive disease and 6870 to have ER-negative disease. Details of included studies are summarized in Online Resource 1. All studies were approved by the relevant ethics committees and all participants gave informed consent (Michailidou et al. 2013).
Candidate gene selection
Candidate genes relevant to the Treg cell and MDSC pathways were identified through a comprehensive literature review in PubMed (DeNardo et al. 2010; DeNardo and Coussens 2007; Driessens et al. 2009; Gabrilovich and Nagaraj 2009; Krieg and Boyman 2009; Mills 2004; Ostrand-Rosenberg 2008; Poschke et al. 2011; Sakaguchi et al. 2013; Sica et al. 2008; Wilczynski and Duechler 2010; Zitvogel et al. 2006; Zou 2005), using the search terms “immunosuppression”/“immunosuppressive”, “regulatory T cells”/“Treg cells”/“FOXP3+ T cells”, “myeloid derived suppressor cells”/“MDSCs”, “immunosurveillance”, and “tumor escape”. The final candidate gene list included 133 immunosuppression-related genes (Online Resource 2). SNPs within 50 kb upstream and downstream of each gene were identified using HapMap CEU genotype data (2015) and dbSNP 126.
SNP association analyses
For the BCAC studies, genotyping was carried out using a custom Illumina iSelect array (iCOGS) designed for the Collaborative Oncological Gene-Environment Study (COGS) project (Michailidou et al. 2013). Of the 211,155 SNPs on the array, 4246 were located within 50 kb of the selected candidate genes. Centralized quality control of genotype data led to the exclusion of 651 SNPs. The exclusion criteria included a call rate less than 95 % in all samples genotyped with iCOGS, minor allele frequency (MAF) less than 0.05 in all samples, evidence of deviation from Hardy–Weinberg equilibrium (HWE) at p value <10−7, and concordance in duplicate samples less than 98 % (Michailidou et al. 2013). A total of 3595 SNPs passed all quality controls and was analyzed.
Per-allele associations with the number of minor alleles were assessed using multiple logistic regression models, adjusted for study, age (at diagnosis for cases or at recruitment for controls) and nine principal components (PCs) derived based on genotyped variants to account for European population substructure. We assessed the associations of SNPs with overall breast cancer risk as primary analyses, and then restricted to ER-positive (26,094 cases and 40,577 controls) and ER-negative subtypes (6870 cases and 40,577 controls) as secondary analyses. Differences in the associations between ER-positive and ER-negative diseases were assessed by case-only analyses, using ER status as the dependent variable. To determine the number of “independent” SNPs for adjustment of multiple testing, we applied the option “--indep-pairwise” in PLINK (Purcell et al. 2007). SNPs were pruned by linkage disequilibrium (LD) of r 2 < 0.2 for a window size of 50 SNPs and step size of 10 SNPs, yielding 689 “independent” SNPs. The significance threshold using Bonferroni correction corresponding to an alpha of 5 % was 7.3 × 10−5.
In order to identify more strongly associated variants, genotypes were imputed for SNPs at the locus for which strongest evidence of association was observed, via a two-stage procedure involving SHAPEIT (Howie et al. 2012) and IMPUTEv2 (Howie et al. 2009), using the 1000 Genomes Project data as the reference panel (Abecasis et al. 2012). Details of the imputation procedure are described elsewhere (Michailidou et al. 2015). Models assessing associations with imputed SNPs were adjusted for 16 PCs based on 1000 Genome imputed data to further improve adjustment for population stratification. To determine independent signals within imputed SNPs at STAT3, we ran a stepwise forward multiple logistic regression model including the most significant genotyped SNP rs1905339 and all imputed SNPs, adjusted for study, age and 16 PCs.
SNP association analyses and case-only analyses were all conducted using SAS 9.3 (Cary, NC, USA). All tests were two-sided.
For multiple associated SNPs located at the same gene, a Microsoft Excel SNP tool created by Chen et al. (2009) and the software HaploView 4.2 (Barrett et al. 2005) were used to examine LD structure between these SNPs. To be able to inspect LD structures and also for gene-level analyses, allele dosages of imputed SNPs had to be converted into the most probable genotypes. Therefore, we categorized the imputed allele dosage between [0, 0.5] as homozygote of the reference allele, the value between [0.5, 1.5] as heterozygote, and the value between [1.5, 2.0] as homozygote of the counted allele. The regional association plot was generated using the online tool LocusZoom (Pruim et al. 2010).
Gene-level and pathway association analyses
Gene-level associations were determined by a subset of PCs, which were derived from a linear combination of SNPs in each gene explaining 80 % of the variation in the joint distribution of all relevant SNPs. Associations with derived PCs were assessed within a logistic regression framework (Biernacka et al. 2012), for overall breast cancer, ER-positive and ER-negative diseases, respectively. Pathway association of the immunosuppression pathway was assessed based on a global test of association by combining the gene-level p values via the Gamma method (Biernacka et al. 2012). For gene-level associations, associations with p value <3.8 × 10−4 (Bonferroni correction) were considered statistically significant. To gain empirical p values for gene-level associations of TGFBR2 and CCND1 as well as for the pathway association, a Monte Carlo procedure was used with up to 1,000,000 randomizations (Biernacka et al. 2012). An exact binomial test based on the results of the single SNPs association analyses was carried out to estimate enrichment of association in the immunosuppression pathway. Gene-level and pathway association analyses were carried out in R (version 3.1.1) using the package ‘GSAgm’ version 1.0.
Haplotype analyses
To follow up the interesting gene associations observed, haplotype analyses were performed to identify potential susceptibility variants. Haplotype frequencies were determined with the use of the estimation maximization (EM) algorithm (Long et al. 1995) implemented in PROC HAPLOTYPE in SAS 9.3 (Cary, NC, USA). Haplotypes with frequency more or equal than 1 % were examined and the most common haplotype was used as the reference. Rare haplotypes with frequency less than 1 % were grouped into one category. Haplotype-specific odds ratios (ORs) and 95 % confidence intervals (CIs) were estimated within a multiple logistic regression framework, adjusted for the same covariates as in the single SNP association analyses. Global p values for association of haplotypes with breast cancer risk were computed using a likelihood ratio test comparing models with and without haplotypes of the gene of interest.
Gene expression analyses
In order to examine whether potential causative genes influence RNA expression in breast tumor tissue, we downloaded RNA sequence level 3 data from The Cancer Genome Atlas (TCGA) (2015). We retrieved the RNA expression level as the form of RNA-Seq by expectation–maximization (RSEM) based on the IlluminaHiSeq_RNASeqV2 array. Gene expression differences in RNA levels between 989 invasive breast cancer tissues and 113 matched normal tissues for four genes of interest (STAT3, PTRF, IL5, and GM-CSF) were analyzed using a two-sided Wilcoxon–Mann–Whiney test. In addition, data from 183 breast tissues in the GTEx (V6) (2015) publically available online databases were evaluated to obtain information on whether the most interesting variants (rs1905339, rs8074296, rs146170568, chr17:40607850:I and rs77942990) were expression quantitative trait loci (eQTL) for any gene. Also, GTEx was queried to obtain information on whether the five variants were eQTL for STAT3 or PTRF.
Functional annotation
To investigate potential regulatory functions of interesting polymorphisms, we used the Encyclopedia of DNA Elements (ENCODE) database through the UCSC Genome Browser as well as Haploreg v4 (Ward and Kellis 2012).
Results
Selected characteristics of the study population are described in Table 1. The controls and breast cancer patients included in this study had comparable mean reference ages of 54.8 and 55.9 years and also the proportion of postmenopausal women was similar (68 % in controls and 69 % in breast cancer patients). The proportion of women indicating a family history of breast cancer in first degree relatives was as expected greater in breast cancer patients (25 %) than in controls (12 %).
Single SNP associations
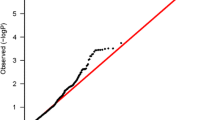
Excluding the known TGFBR2 and CCND1 breast cancer susceptibility loci, the quantile–quantile (QQ) plot for associations with overall breast cancer risk for the genotyped SNPs of the other candidate genes indicated deviation from expected p values and thus evidence of further SNPs associated with breast cancer risk (Online Resource 3). Genetic associations with overall breast cancer risk for all assessed 3595 SNPs are summarized in Online Resource 4.
Four independent genotyped SNPs (LD r 2 < 0.3) were significantly associated with breast cancer risk at p value <7.3 × 10−5, accounting for the multiple comparisons (Table 2). The four significant SNPs were located in or near TGFBR2, STAT3 and CCND1. Since TGFBR2 and CCND1 have been identified as breast cancer susceptibility loci in previous studies (French et al. 2013; Michailidou et al. 2013; Rhie et al. 2013), we focused on the association of the SNP at STAT3. The variant rs1905339 (A>G) at STAT3 was positively associated with overall breast cancer risk (per allele odds ratio (OR) 1.05, 95 % confidence interval (CI) 1.03–1.08, p value = 1.4 × 10−6). It showed similar associations with ER-positive and ER-negative cancers (Online Resource 5). We did not observe further SNPs that were significantly associated with ER-positive or ER-negative disease (data not shown).
To identify additional susceptibility variants at STAT3, we further investigated 707 SNPs that were well-imputed (imputation accuracy r 2 > 0.3) and with MAF >0.01 spanning a ±50 kb window around STAT3. Seven independent signals at STAT3 were found through the stepwise forward selection procedure. The genotyped SNP rs1905339 was not selected. The imputed SNP rs8074296 (A>G), which was in high LD with rs1905339 (r 2 = 0.99), showed a comparable OR for the association with overall breast cancer risk with a more extreme p value (per allele OR 1.05, 95 % CI 1.03–1.08, p value = 8.6 × 10−7, Table 3). A second imputed SNP rs146170568 (C>T), associated with a per allele OR of 1.32 (95 % CI 1.16–1.50, p value = 2.1 × 10−5), was still strongly associated at a p value of 3.2 × 10−4 after accounting for rs8074296 (Table 3). None of the independently associated imputed SNPs besides rs8074296 were correlated with rs1905339 or with each other (r 2 ≤ 0.01, Fig. 1). As rs8074296 and rs1905339 are located closer to PTRF than to STAT3, we additionally analyzed data of 178 imputed variants located within ±50 kb of PTRF. Associations of most additional variants in the PTRF region with breast cancer risk were attenuated in analyses conditioning on rs8074296 (Table 4). The variants chr17:40607850:I and rs77942990 still showed a strong association with breast cancer risk (per allele OR 1.09, 95 % CI 1.04–1.15, p value = 0.0005; and per allele OR 1.09, 95 % CI 1.04–1.15, p value = 0.0007, respectively). These two variants were also not in LD with rs8074296 (r 2 = 0.09 and 0.07, respectively) while all other variants in Table 4 were at least in moderate LD with rs8074296 (r 2 ≥ 0.46, Online Resource 6). The LD plot (Online Resource 6) also shows that chr17:40607850:I and rs77942990 are in high LD (r 2 = 0.83). A regional association plot for the genotyped SNP rs1905339 and all 885 imputed SNPs within ±50 kb of STAT3 and PTRF included in this analysis is shown in Fig. 2. Associations of SNPs shown in Table 3 as well as associations of chr17:40607850:I and rs77942990 with breast cancer risk were not significantly heterogeneous between studies (all p values for heterogeneity >0.1); forest plots can be found in Online Resource 7 to 16.

Linkage disequilibrium plot showing r 2 values and color schemes for the genotyped SNP rs1905339 and seven independent imputed SNPs as well as imputed SNP rs181888151 within ±50 kb of STAT3. The linkage disequilibrium (LD) plot shows that SNP rs1905339 is in strong LD with the imputed SNP rs8074296 (r 2 = 0.99), and independent of the other six imputed SNPs (r 2 ≤ 0.01) at STAT3. LD was estimated based on control data

Regional association plot for the genotyped SNP rs1905339 and 885 imputed SNPs within ±50 kb of STAT3 and PTRF. Each dot represents an SNP. The color of each dot reflects the extent of linkage disequilibrium (r 2) with SNP rs1032070 (in purple diamond). Genomic positions of SNPs were plotted based on hg19/1000 Genomes Mar 2012 European. Association is represented at the −log10 scale. cM/Mb centiMorgans/megabase
Gene-level and pathway associations
Gene-level associations with risks of overall breast cancer, ER-positive and ER-negative diseases, respectively, for the 133 candidate genes in the immunosuppression pathway are summarized in Online Resource 17. TGFBR2 and CCND1 showed significant associations with overall breast cancer risk (p value <10−6 and 3.0 × 10−4, respectively). In addition, IL5 and GM-CSF may be further potential susceptibility loci of breast cancer (p value = 1.0 × 10−3 and 7.0 × 10−3, respectively). STAT3 showed a less significant association with overall breast cancer risk (p value = 0.033). The immunosuppression pathway as a whole yielded a significant association with overall breast cancer risk (p value <10−6). Similar gene-level and pathway associations were found for ER-positive but not for ER-negative breast cancer (Online Resource 17). We found significant enrichment of association in the immunosuppression pathway based on the results of the single SNPs association analyses (313 of 3595 tests significant at α = 0.05, exact binomial test p value = 2.2 × 10−16).
Haplotype analyses
Despite the evidence for a possible role of IL5 and GM-CSF in breast cancer susceptibility from the gene-level analysis, no individual SNPs at IL5 or GM-CSF yielded significant genetic associations. To identify potential susceptibility haplotypes, haplotype-specific associations were assessed based on seven SNPs in or near IL5 (rs4143832, rs2079103, rs2706399, rs743562, rs739719, rs2069812 and rs2244012) and nine SNPs in or near GM-CSF (rs11575022, rs2069616, rs25881, rs25882, rs25883, rs27349, rs27438, rs40401 and rs743564). The LD structures for these SNPs at IL5 and GM-CSF are shown in Online Resource 18 and 19, respectively. In our study sample of women of European ancestry, 11 and 7 common haplotypes with frequency >1 % were observed at IL5 and GM-CSF, respectively. The haplotype AAAACGG in IL5 was associated with a decreased overall breast cancer risk (OR 0.96, 95 % CI 0.93–0.99, p value = 5.0 × 10−3, Table 5). In GM-CSF, the haplotype AAGAGCGAA was also associated with a decreased overall breast cancer risk (OR 0.92, 95 % CI 0.87–0.96, p value = 2.7 × 10−4, Table 6). The global p value for haplotype association was significant for both IL5 (p value = 0.005) and GM-CSF (p value = 0.007).
Gene expression analyses
Using TCGA RNA sequencing level 3 data, we found that RNA expression levels of STAT3 and IL5 were significantly higher in 113 normal tissue samples compared to 989 breast tumor samples (p value = 1.3 × 10−3 and 7.0 × 10−4, respectively, Online Resources 20 and 21), while overall expression of IL5 was low in both tissues. Also expression levels of PTRF were significantly higher in normal tissue compared to tumor tissue samples (p value ≤0.0001, Online Resource 22). GM-CSF expression was very low and did not differ between breast tumor samples and normal tissue samples (p value = 0.49, Online Resource 23). Among 183 mammary tissues in the GTEx database, SNPs rs1905339, rs8074296 and rs77942990 were not significantly correlated with STAT3 (p values = 0.36, 0.36, and 0.2, respectively; Online Resource 24 to 26) or PTRF expression (p values = 0.4, 0.4, and 0.39 Online Resource 27 to 29). The SNPs rs1905339 and rs8074296 were significant eQTL for TUBG2 (both p values = 9.9 × 10−7, Online Resource 30 and 31). The STAT3/PTRF variants rs146170568 and chr17:40607850:I were not available in the GTEx database.
Discussion
Our comprehensive examination of associations between polymorphisms in the immunosuppression pathway genes and breast cancer risk revealed that STAT3, IL5, and GM-CSF may play a role in overall breast cancer susceptibility among women of European ancestry.
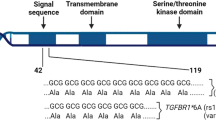
The in silico functional analysis revealed that within a ±50 kb window of STAT3, several polymorphisms are located in regulatory regions that could actively affect DNA transcription (Fig. 3). The SNP rs181888151, which is in complete LD with rs146170568 (r 2 = 1) but independent of rs1905339 (r 2 = 0.01, Fig. 1) was significantly associated with increased risk for overall breast cancer (per allele OR 1.31, 95 % CI 1.16–1.49, p value = 2.8 × 10−5). Together with a further independently associated imputed SNP rs141732716, these polymorphisms reside in strong DNase I hypersensitivity and transcription regulatory sites (Fig. 3). This suggests that they may be functional polymorphisms, but further experimental work is required for confirmation.

UCSC genome browser graphic for SNPs at the STAT3/PTRF region. The UCSC genome browser graphic shows functional annotations for the SNPs rs1905339 (red), correlated SNPs (r 2 > 0.80, green), as well as the other independent imputed SNPs (black) in or near the STAT3/PTRF region
STAT3 encodes the signal transducer and activator of transcription 3, which is a member of the STAT protein family. Activated by corresponding cytokines or growth factors, STAT3 can be phosphorylated and translocate into the cell nucleus, acting as a transcription activator. In addition, STAT3 plays a key role in regulating immune response in the tumor microenvironment (Yu et al. 2009). STAT3 signaling is required for immunosuppressive and tumor-promoting functions of MDSCs (Cheng et al. 2003, 2008; Kortylewski et al. 2005, 2009; Kujawski et al. 2008; Ostrand-Rosenberg and Sinha 2009; Yu et al. 2009), as well as for Treg cell expansion (Kortylewski et al. 2005, 2009; Matsumura et al. 2007). STAT3 has been reported in several previous genome-wide association studies (GWAS) to be associated with immune relevant diseases such as Crohn’s disease (Barrett et al. 2008; Franke et al. 2008; Yamazaki et al. 2013), inflammatory bowel disease (Jostins et al. 2012), and multiple sclerosis (Jakkula et al. 2010; Patsopoulos et al. 2011; Sawcer et al. 2011). Additionally, expression of STAT3 was suggested to be enriched in triple-negative breast cancer, and negatively associated with lymph node involvement and breast tumor stage in a study based on an in silico network approach (Liu et al. 2012b). However, the association of rs1905339 with triple-negative breast cancer risk in our study (N triple-negative breast cancer = 2600) was similar and not stronger compared to the association observed for overall breast cancer risk (per allele OR 1.06, 95 % CI 0.99–1.14, p value = 0.11).
The genotyped SNP rs1905339 is also located at 7 kb 5′ of PTRF, which encodes the polymerase I and transcript release factor, and is not known to be directly involved in immunosuppression. In addition, two independently associated imputed SNPs rs8074296 and rs12952342 (r 2 = 0.99 and 0 with rs1905339, respectively, Fig. 1) are located at 8 kb 5′ and 0.8 kb 3′ of PTRF, respectively (Fig. 3). PTRF is known to contribute to the formation of caveolae, small membrane caves involved in cell signaling, lipid regulation, and endocytosis (Chadda and Mayor 2008). Recently, down-regulation of PTRF was observed in breast cancer cell lines and breast tumor tissue, suggesting that PTRF expression might be an indicator for breast cancer progression (Bai et al. 2012). The SNPs rs1905339 and rs8074296 were also found to be eQTL for TUBG2 (tubulin, gamma 2) in the GTEx database, the expression of TUBG2 decreased with each variant allele (Online Resources 30 and 31, respectively). TUBG2 encodes γ-tubulin, a protein required for the formation and polar orientation of microtubules in cells. It is currently unknown, whether TUBG2 plays a role in breast cancer development or progression.
The other two potential susceptibility loci, IL5 and GM-CSF, are both located in a known cytokine gene cluster at 5q31. IL5 encodes interleukin 5, a cytokine secreted by CD4+ T helper 2 cells (Mills 2004; Parker 1993). IL5 is a growth and differentiation factor for both B cells and eosinophils, triggering eosinophil- and B cell-dependent immune response (Mills 2004; Parker 1993). GM-CSF encodes granulocyte–macrophage colony stimulating factor, a cytokine that controls differentiation and function of granulocytes and macrophages. GM-CSF is also a MDSC- inducing and activating factor in the bone marrow (Ostrand-Rosenberg and Sinha 2009; Serafini et al. 2004). In the tumor microenvironment, GM-CSF is the cytokine for dendritic cell differentiation and function, and it is often found to be underexpressed (Zou 2005). Additionally, 5q31 has been found to be a susceptibility locus for rheumatoid arthritis (Okada et al. 2012, 2014) and inflammatory bowel disease (Jostins et al. 2012).
Immunosuppression is a complex network with plenty of contributors, including transcription factors (e.g., STAT3), as well as immune mediating cytokines (e.g., IL5 and GM-CSF). Results of this analysis indicate that genetic variation in different components of the immunosuppression pathway may be susceptibility loci of breast cancer among women of European ancestry.
The main strengths of the present analysis were its large sample size, the uniform genotyping procedures and centralized quality controls used. The imputation of genotypes in the most interesting susceptibility loci provided an opportunity to identify more strongly associated variants. Assessments of gene-level associations also provided evidence for additional putative susceptibility loci. A limitation was the lack of an independent sample to replicate the observed associations; this will be feasible in the future using new studies participating in the BCAC. Further functional studies are still needed to identify causal variants and to investigate the underlying biological mechanisms.
Conclusions
Overall, our data provide strong evidence that common variation in the immunosuppression pathway is associated with breast cancer susceptibility. The strongest candidates for mediating this association were STAT3, IL5, and GM-CSF, but we cannot exclude the possibility of multiple alleles each with effects too small to confirm.
Abbreviations
- BCAC:
-
Breast Cancer Association Consortium
- CCND1:
-
Cyclin D1
- CI:
-
Confidence interval
- COGS:
-
Collaborative Oncological Gene-Environment Study
- DNA:
-
Deoxyribonucleic acid
- GM-CSF:
-
Granulocyte-macrophage colony stimulating factor
- EM:
-
Estimation maximization
- ENCODE:
-
Encyclopedia of DNA elements
- eQTL:
-
Expression quantitative trait loci
- ER:
-
Estrogen receptor
- GWAS:
-
Genome-wide association study
- HWE:
-
Hardy–Weinberg equilibrium
- IL5:
-
Interleukin 5
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MDSCs:
-
Myeloid-derived suppressor cells
- OR:
-
Odds ratio
- PCs:
-
Principal components
- PTRF:
-
Polymerase I and transcript release factor
- QQ:
-
Quantile–quantile
- RSEM:
-
RNA-Seq by expectation-maximization
- SD:
-
Standard deviation
- SNPs:
-
Single nucleotide polymorphisms
- STAT3:
-
Signal transducer and activator of transcription 3
- TCGA:
-
The Cancer Genome Atlas
- TGFBR2:
-
Transforming growth factor beta receptor II
- Treg cells:
-
Regulatory T cells
- TUBG2:
-
Tubulin, gamma 2
References
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. doi:10.1038/nature11632
Aliper AM, Frieden-Korovkina VP, Buzdin A, Roumiantsev SA, Zhavoronkov A (2014) Interactome analysis of myeloid-derived suppressor cells in murine models of colon and breast cancer. Oncotarget 5:11345–11353
Bai L, Deng X, Li Q, Wang M, An W, Deli A, Gao Z, Xie Y, Dai Y, Cong YS (2012) Down-regulation of the cavin family proteins in breast cancer. J Cell Biochem 113:322–328. doi:10.1002/jcb.23358
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265. doi:10.1093/bioinformatics/bth457
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40:955–962. doi:10.1038/ng.175
Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH (2006) Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 24:5373–5380. doi:10.1200/jco.2006.05.9584
Biernacka JM, Jenkins GD, Wang L, Moyer AM, Fridley BL (2012) Use of the gamma method for self-contained gene-set analysis of SNP data. Eur J Hum Genet 20:565–571. doi:10.1038/ejhg.2011.236
Breast Cancer Association Consortium. http://www.apps.ccge.medschl.cam.ac.uk/consortia/bcac/. Accessed 19 May 2015
Chadda R, Mayor S (2008) PTRF triggers a cave in. Cell 132:23–24. doi:10.1016/j.cell.2007.12.021
Chang WC, Li CH, Huang SC, Chang DY, Chou LY, Sheu BC (2010) Clinical significance of regulatory T cells and CD8+ effector populations in patients with human endometrial carcinoma. Cancer 116:5777–5788. doi:10.1002/cncr.25371
Chen B, Wilkening S, Drechsel M, Hemminki K (2009) SNP_tools: a compact tool package for analysis and conversion of genotype data for MS-Excel. BMC Res Note 2:214. doi:10.1186/1756-0500-2-214
Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J, Kerr WG, Takeda K, Akira S, Schoenberger SP, Yu H, Jove R, Sotomayor EM (2003) A critical role for Stat3 signaling in immune tolerance. Immunity 19:425–436
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205:2235–2249. doi:10.1084/jem.20080132
DeNardo DG, Coussens LM (2007) Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res 9:212. doi:10.1186/bcr1746
DeNardo DG, Andreu P, Coussens LM (2010) Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev 29:309–316. doi:10.1007/s10555-010-9223-6
Driessens G, Kline J, Gajewski TF (2009) Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev 229:126–144. doi:10.1111/j.1600-065X.2009.00771.x
Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, Schuldt D, Nikolaus S, Rosenstiel P, Krawczak M, Schreiber S (2008) Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet 40:713–715. doi:10.1038/ng.148
French JD, Ghoussaini M, Edwards SL, Meyer KB, Michailidou K, Ahmed S, Khan S, Maranian MJ, O’Reilly M, Hillman KM, Betts JA, Carroll T, Bailey PJ, Dicks E, Beesley J, Tyrer J, Maia AT, Beck A, Knoblauch NW, Chen C, Kraft P, Barnes D, Gonzalez-Neira A, Alonso MR, Herrero D, Tessier DC, Vincent D, Bacot F, Luccarini C, Baynes C, Conroy D, Dennis J, Bolla MK, Wang Q, Hopper JL, Southey MC, Schmidt MK, Broeks A, Verhoef S, Cornelissen S, Muir K, Lophatananon A, Stewart-Brown S, Siriwanarangsan P, Fasching PA, Loehberg CR, Ekici AB, Beckmann MW, Peto J, dos Santos Silva I, Johnson N, Aitken Z, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Guenel P, Truong T, Laurent-Puig P, Menegaux F, Bojesen SE, Nordestgaard BG, Nielsen SF, Flyger H, Milne RL, Zamora MP, Arias Perez JI, Benitez J, Anton-Culver H, Brenner H, Muller H, Arndt V, Stegmaier C, Meindl A, Lichtner P, Schmutzler RK, Engel C, Brauch H, Hamann U, Justenhoven C, Aaltonen K, Heikkila P, Aittomaki K, Blomqvist C, Matsuo K, Ito H, Iwata H, Sueta A, Bogdanova NV, Antonenkova NN, Dork T, Lindblom A, Margolin S, Mannermaa A, Kataja V, Kosma VM et al (2013) Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am J Hum Genet 92:489–503. doi:10.1016/j.ajhg.2013.01.002
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. doi:10.1038/nri2506
Gad E, Rastetter L, Slota M, Koehnlein M, Treuting PM, Dang Y, Stanton S, Disis ML (2014) Natural history of tumor growth and immune modulation in common spontaneous murine mammary tumor models. Breast Cancer Res Treat 148:501–510. doi:10.1007/s10549-014-3199-9
GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. International Agency for Research on Cancer. http://www.globocan.iarc.fr/Default.aspx. Accessed 9 Apr 2015
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. doi:10.1016/j.cell.2011.02.013
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529. doi:10.1371/journal.pgen.1000529
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44:955–959. doi:10.1038/ng.2354
International HapMap Project. http://www.hapmap.org/. Accessed 26 Mar 2015
Jakkula E, Leppa V, Sulonen AM, Varilo T, Kallio S, Kemppinen A, Purcell S, Koivisto K, Tienari P, Sumelahti ML, Elovaara I, Pirttila T, Reunanen M, Aromaa A, Oturai AB, Sondergaard HB, Harbo HF, Mero IL, Gabriel SB, Mirel DB, Hauser SL, Kappos L, Polman C, De Jager PL, Hafler DA, Daly MJ, Palotie A, Saarela J, Peltonen L (2010) Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am J Hum Genet 86:285–291. doi:10.1016/j.ajhg.2010.01.017
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE et al (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491:119–124. doi:10.1038/nature11582
Kim ST, Jeong H, Woo OH, Seo JH, Kim A, Lee ES, Shin SW, Kim YH, Kim JS, Park KH (2013) Tumor-infiltrating lymphocytes, tumor characteristics, and recurrence in patients with early breast cancer. Am J Clin Oncol 36:224–231. doi:10.1097/COC.0b013e3182467d90
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11:1314–1321. doi:10.1038/nm1325
Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, Drake C, Pardoll D, Yu H (2009) Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 15:114–123. doi:10.1016/j.ccr.2008.12.018
Krieg C, Boyman O (2009) The role of chemokines in cancer immune surveillance by the adaptive immune system. Semin Cancer Biol 19:76–83. doi:10.1016/j.semcancer.2008.10.011
Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H (2008) Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 118:3367–3377. doi:10.1172/jci35213
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ (2013) The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 138:105–115. doi:10.1111/imm.12036
Liu F, Li Y, Ren M, Zhang X, Guo X, Lang R, Gu F, Fu L (2012a) Peritumoral FOXP3(+) regulatory T cell is sensitive to chemotherapy while intratumoral FOXP3(+) regulatory T cell is prognostic predictor of breast cancer patients. Breast Cancer Res Treat 135:459–467. doi:10.1007/s10549-012-2132-3
Liu LY, Chang LY, Kuo WH, Hwa HL, Lin YS, Huang SF, Chen CN, Chang KJ, Hsieh FJ (2012b) Major Functional Transcriptome of an Inferred Center Regulator of an ER(-) Breast Cancer Model System. Cancer Inform 11:87–111. doi:10.4137/cin.s8633
Long JC, Williams RC, Urbanek M (1995) An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet 56:799–810
Matsumura Y, Kobayashi T, Ichiyama K, Yoshida R, Hashimoto M, Takimoto T, Tanaka K, Chinen T, Shichita T, Wyss-Coray T, Sato K, Yoshimura A (2007) Selective expansion of foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J Immunol 179:2170–2179
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J, Bojesen SE, Bolla MK, Wang Q, Dicks E, Lee A, Turnbull C, Rahman N, Fletcher O, Peto J, Gibson L, Dos Santos Silva I, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Czene K, Irwanto A, Liu J, Waisfisz Q, Meijers-Heijboer H, Adank M, van der Luijt RB, Hein R, Dahmen N, Beckman L, Meindl A, Schmutzler RK, Muller-Myhsok B, Lichtner P, Hopper JL, Southey MC, Makalic E, Schmidt DF, Uitterlinden AG, Hofman A, Hunter DJ, Chanock SJ, Vincent D, Bacot F, Tessier DC, Canisius S, Wessels LF, Haiman CA, Shah M, Luben R, Brown J, Luccarini C, Schoof N, Humphreys K, Li J, Nordestgaard BG, Nielsen SF, Flyger H, Couch FJ, Wang X, Vachon C, Stevens KN, Lambrechts D, Moisse M, Paridaens R, Christiaens MR, Rudolph A, Nickels S, Flesch-Janys D, Johnson N, Aitken Z, Aaltonen K, Heikkinen T, Broeks A, Veer LJ, van der Schoot CE, Guenel P, Truong T, Laurent-Puig P, Menegaux F, Marme F, Schneeweiss A, Sohn C, Burwinkel B, Zamora MP, Perez JI, Pita G, Alonso MR, Cox A, Brock IW, Cross SS, Reed MW, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N, Henderson BE et al (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45:353–361. doi:10.1038/ng.2563 (361e1-2)
Michailidou K, Beesley J, Lindstrom S, Canisius S, Dennis J, Lush MJ, Maranian MJ, Bolla MK, Wang Q, Shah M, Perkins BJ, Czene K, Eriksson M, Darabi H, Brand JS, Bojesen SE, Nordestgaard BG, Flyger H, Nielsen SF, Rahman N, Turnbull C, Fletcher O, Peto J, Gibson L, Dos-Santos-Silva I, Chang-Claude J, Flesch-Janys D, Rudolph A, Eilber U, Behrens S, Nevanlinna H, Muranen TA, Aittomaki K, Blomqvist C, Khan S, Aaltonen K, Ahsan H, Kibriya MG, Whittemore AS, John EM, Malone KE, Gammon MD, Santella RM, Ursin G, Makalic E, Schmidt DF, Casey G, Hunter DJ, Gapstur SM, Gaudet MM, Diver WR, Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Berg CD, Chanock SJ, Figueroa J, Hoover RN, Lambrechts D, Neven P, Wildiers H, van Limbergen E, Schmidt MK, Broeks A, Verhoef S, Cornelissen S, Couch FJ, Olson JE, Hallberg E, Vachon C, Waisfisz Q, Meijers-Heijboer H, Adank MA, van der Luijt RB, Li J, Liu J, Humphreys K, Kang D, Choi JY, Park SK, Yoo KY, Matsuo K, Ito H, Iwata H, Tajima K, Guenel P, Truong T, Mulot C, Sanchez M, Burwinkel B, Marme F, Surowy H, Sohn C, Wu AH, Tseng CC, Van Den Berg D, Stram DO, Gonzalez-Neira A, Benitez J et al (2015) Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat Genet 47:373–380. doi:10.1038/ng.3242
Michel S, Benner A, Tariverdian M, Wentzensen N, Hoefler P, Pommerencke T, Grabe N, von Knebel Doeberitz M, Kloor M (2008) High density of FOXP3-positive T cells infiltrating colorectal cancers with microsatellite instability. Br J Cancer 99:1867–1873. doi:10.1038/sj.bjc.6604756
Mills KH (2004) Regulatory T cells: friend or foe in immunity to infection? Nat Rev Immunol 4:841–855. doi:10.1038/nri1485
Okada Y, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Kawaguchi T, Stahl EA, Kurreeman FA, Nishida N, Ohmiya H, Myouzen K, Takahashi M, Sawada T, Nishioka Y, Yukioka M, Matsubara T, Wakitani S, Teshima R, Tohma S, Takasugi K, Shimada K, Murasawa A, Honjo S, Matsuo K, Tanaka H, Tajima K, Suzuki T, Iwamoto T, Kawamura Y, Tanii H, Okazaki Y, Sasaki T, Gregersen PK, Padyukov L, Worthington J, Siminovitch KA, Lathrop M, Taniguchi A, Takahashi A, Tokunaga K, Kubo M, Nakamura Y, Kamatani N, Mimori T, Plenge RM, Yamanaka H, Momohara S, Yamada R, Matsuda F, Yamamoto K (2012) Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet 44:511–516. doi:10.1038/ng.2231
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, Graham RR, Manoharan A, Ortmann W, Bhangale T, Denny JC, Carroll RJ, Eyler AE, Greenberg JD, Kremer JM, Pappas DA, Jiang L, Yin J, Ye L, Su DF, Yang J, Xie G, Keystone E, Westra HJ, Esko T, Metspalu A, Zhou X, Gupta N, Mirel D, Stahl EA, Diogo D, Cui J, Liao K, Guo MH, Myouzen K, Kawaguchi T, Coenen MJ, van Riel PL, van de Laar MA, Guchelaar HJ, Huizinga TW, Dieude P, Mariette X, Bridges SL Jr, Zhernakova A, Toes RE, Tak PP, Miceli-Richard C, Bang SY, Lee HS, Martin J, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Rantapaa-Dahlqvist S, Arlestig L, Choi HK, Kamatani Y, Galan P, Lathrop M, Eyre S, Bowes J, Barton A, de Vries N, Moreland LW, Criswell LA, Karlson EW, Taniguchi A, Yamada R, Kubo M, Liu JS, Bae SC, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Raychaudhuri S, Stranger BE, De Jager PL, Franke L, Visscher PM, Brown MA, Yamanaka H, Mimori T, Takahashi A, Xu H, Behrens TW, Siminovitch KA, Momohara S, Matsuda F, Yamamoto K, Plenge RM (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506:376–381. doi:10.1038/nature12873
Ostrand-Rosenberg S (2008) Immune surveillance: a balance between protumor and antitumor immunity. Curr Opin Genet Dev 18:11–18. doi:10.1016/j.gde.2007.12.007
Ostrand-Rosenberg S, Sinha P (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 182:4499–4506. doi:10.4049/jimmunol.0802740
Parker DC (1993) T cell-dependent B cell activation. Annu Rev Immunol 11:331–360. doi:10.1146/annurev.iy.11.040193.001555
Patsopoulos NA, Esposito F, Reischl J, Lehr S, Bauer D, Heubach J, Sandbrink R, Pohl C, Edan G, Kappos L, Miller D, Montalban J, Polman CH, Freedman MS, Hartung HP, Arnason BG, Comi G, Cook S, Filippi M, Goodin DS, Jeffery D, O’Connor P, Ebers GC, Langdon D, Reder AT, Traboulsee A, Zipp F, Schimrigk S, Hillert J, Bahlo M, Booth DR, Broadley S, Brown MA, Browning BL, Browning SR, Butzkueven H, Carroll WM, Chapman C, Foote SJ, Griffiths L, Kermode AG, Kilpatrick TJ, Lechner-Scott J, Marriott M, Mason D, Moscato P, Heard RN, Pender MP, Perreau VM, Perera D, Rubio JP, Scott RJ, Slee M, Stankovich J, Stewart GJ, Taylor BV, Tubridy N, Willoughby E, Wiley J, Matthews P, Boneschi FM, Compston A, Haines J, Hauser SL, McCauley J, Ivinson A, Oksenberg JR, Pericak-Vance M, Sawcer SJ, De Jager PL, Hafler DA, de Bakker PI (2011) Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol 70:897–912. doi:10.1002/ana.22609
Poschke I, Mougiakakos D, Kiessling R (2011) Camouflage and sabotage: tumor escape from the immune system. Cancer Immunol Immunother 60:1161–1171. doi:10.1007/s00262-011-1012-8
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26:2336–2337. doi:10.1093/bioinformatics/btq419
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. doi:10.1086/519795
Reisfeld RA (2013) The tumor microenvironment: a target for combination therapy of breast cancer. Crit Rev Oncog 18:115–133
Rhie SK, Coetzee SG, Noushmehr H, Yan C, Kim JM, Haiman CA, Coetzee GA (2013) Comprehensive functional annotation of seventy-one breast cancer risk Loci. PLoS One 8:e63925. doi:10.1371/journal.pone.0063925
Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H (2013) The plasticity and stability of regulatory T cells. Nat Rev Immunol 13:461–467. doi:10.1038/nri3464
Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D’Alfonso S, Blackburn H, Martinelli Boneschi F, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget-Darpoux F, Clysters K, Comi G, Cossburn M, Cournu-Rebeix I, Cox MB, Cozen W, Cree BA, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PI, Debouverie M, D’Hooghe MB, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F et al (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476:214–219. doi:10.1038/nature10251
Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I (2004) High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res 64:6337–6343. doi:10.1158/0008-5472.can-04-0757
Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A (2008) Macrophage polarization in tumour progression. Semin Cancer Biol 18:349–355. doi:10.1016/j.semcancer.2008.03.004
The Cancer Genome Atlas. http://www.cancergenome.nih.gov/. Accessed 1 Apr 2015
The Genotype-Tissue Expression Portal. http://www.gtexportal.org/home/. Accessed 19 Oct 2015
UCSC Genome Browser. https://www.genome-euro.ucsc.edu/cgi-bin/hgGateway?redirect=manual&source=genome.ucsc.edu. Accessed 19 Oct 2015
Ward LD, Kellis M (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acid Res 40:D930–D934. doi:10.1093/nar/gkr917
Watanabe Y, Kinoshita A, Yamada T, Ohta T, Kishino T, Matsumoto N, Ishikawa M, Niikawa N, Yoshiura K (2002) A catalog of 106 single-nucleotide polymorphisms (SNPs) and 11 other types of variations in genes for transforming growth factor-beta1 (TGF-beta1) and its signaling pathway. J Hum Genet 47:478–483. doi:10.1007/s100380200069
Wilczynski JR, Duechler M (2010) How do tumors actively escape from host immunosurveillance? Arch Immunol Ther Exp (Warsz) 58:435–448. doi:10.1007/s00005-010-0102-1
Yamazaki K, Umeno J, Takahashi A, Hirano A, Johnson TA, Kumasaka N, Morizono T, Hosono N, Kawaguchi T, Takazoe M, Yamada T, Suzuki Y, Tanaka H, Motoya S, Hosokawa M, Arimura Y, Shinomura Y, Matsui T, Matsumoto T, Iida M, Tsunoda T, Nakamura Y, Kamatani N, Kubo M (2013) A genome-wide association study identifies 2 susceptibility Loci for Crohn’s disease in a Japanese population. Gastroenterology 144:781–788. doi:10.1053/j.gastro.2012.12.021
Yang L, Pang Y, Moses HL (2010) TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol 31:220–227. doi:10.1016/j.it.2010.04.002
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809. doi:10.1038/nrc2734
Zitvogel L, Tesniere A, Kroemer G (2006) Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol 6:715–727. doi:10.1038/nri1936
Zou W (2005) Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 5:263–274. doi:10.1038/nrc1586
Acknowledgments
We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians, and administrative staff who have enabled this work to be carried out. This analysis would not have been possible without the contributions of the following: Per Hall (COGS); Douglas F. Easton, Paul Pharoah, Kyriaki Michailidou, Manjeet K. Bolla, Qin Wang (BCAC), Andrew Berchuck (OCAC), Rosalind A. Eeles, Douglas F. Easton, Ali Amin Al Olama, Zsofia Kote-Jarai, Sara Benlloch (PRACTICAL), Georgia Chenevix-Trench, Antonis Antoniou, Lesley McGuffog, Fergus Couch and Ken Offit (CIMBA), Joe Dennis, Alison M. Dunning, Andrew Lee, and Ed Dicks, Craig Luccarini and the staff of the Centre for Genetic Epidemiology Laboratory, Javier Benitez, Anna Gonzalez-Neira and the staff of the CNIO genotyping unit, Jacques Simard and Daniel C. Tessier, Francois Bacot, Daniel Vincent, Sylvie LaBoissière and Frederic Robidoux and the staff of the McGill University and Génome Québec Innovation Centre, Stig E. Bojesen, Sune F. Nielsen, Borge G. Nordestgaard, and the staff of the Copenhagen DNA laboratory, and Julie M. Cunningham, Sharon A. Windebank, Christopher A. Hilker, Jeffrey Meyer and the staff of Mayo Clinic Genotyping Core Facility. ABCFS would like to thank Maggie Angelakos, Judi Maskiell, and Gillian Dite. ABCS would like to thank Sanquin Amsterdam, the Netherlands. BBCS thanks Eileen Williams, Elaine Ryder-Mills, and Kara Sargus. BIGGS thanks Niall McInerney, Gabrielle Colleran, Andrew Rowan, and Angela Jones. BSUCH would like to thank Peter Bugert and Medical Faculty Mannheim. CGPS thanks Staff and participants of the Copenhagen General Population Study, as well as excellent technical assistance from Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, and Dorthe Kjeldgård Hansen. CNIO-BCS would like to thank Guillermo Pita, Charo Alonso, Daniel Herrero, Nuria Álvarez, Pilar Zamora, Primitiva Menendez, and the Human Genotyping-CEGEN Unit. CTS would like to thank the CTS Steering Committee including Leslie Bernstein, Susan Neuhausen, James Lacey, Sophia Wang, Huiyan Ma, Yani Lu, and Jessica Clague DeHart at the Beckman Research Institute of City of Hope, Dennis Deapen, Rich Pinder, Eunjung Lee, and Fred Schumacher at the University of Southern California, Pam Horn-Ross, Peggy Reynolds, Christina Clarke Dur and David Nelson at the Cancer Prevention Institute of California, and Hoda Anton-Culver, Argyrios Ziogas, and Hannah Park at the University of California Irvine. ESTHER thanks Hartwig Ziegler, Christa Stegmaier, Sonja Wolf, and Volker Hermann. GC-HBOC thanks Stefanie Engert, Heide Hellebrand, and Sandra Kröber. GENICA would like to thank the GENICA Network, including Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, and University of Tübingen, Germany (HB, Wing-Yee Lo, Christina Justenhoven), German Cancer Consortium (DKTK) and Deutsches Krebsforschungszentrum (DKFZ) (HB), Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany (Yon-Dschun Ko, Christian Baisch), Institute of Pathology, University of Bonn, Germany (Hans-Peter Fischer), Molecular Genetics of Breast Cancer, DKFZ, Heidelberg, Germany (UH), Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany (Thomas Brüning, Beate Pesch, Sylvia Rabstein, Anne Lotz), and Institute of Occupational Medicine and Maritime Medicine, University Medical Center Hamburg-Eppendorf, Germany (Volker Harth). HEBCS would like to thank Kirsimari Aaltonen, Karl von Smitten, Sofia Khan, Tuomas Heikkinen, and Irja Erkkilä. HMBCS would like to thank Peter Hillemanns, Hans Christiansen, and Johann H. Karstens. KBCP thanks Eija Myöhänen and Helena Kemiläinen. LMBC thanks Gilian Peuteman, Dominiek Smeets, Thomas Van Brussel, and Kathleen Corthouts. MARIE would like to thank Petra Seibold, Judith Heinz, Nadia Obi, Alina Vrieling, Muhabbet Celik, Til Olchers, and Stefan Nickels. MBCSG thanks Siranoush Manoukian, Bernard Peissel and Daniela Zaffaroni at the Fondazione IRCCS Istituto Nazionale dei Tumori (INT), Monica Barile and Irene Feroce at the Istituto Europeo di Oncologia (IEO), and the personnel of the Cogentech Cancer Genetic Test Laboratory. MTLGEBCS would like to thank Martine Tranchant at the CHU de Québec Research Center, Marie-France Valois, Annie Turgeon and Lea Heguy at the McGill University Health Center, Royal Victoria Hospital, McGill University for DNA extraction, sample management and skillful technical assistance, and J.S. who is the Chairholder of the Canada Research Chair in Oncogenetics. NBCS would like to thank Dr. Kristine Kleivi, PhD (K.G. Jebsen Centre for Breast Cancer Research, Institute of Clinical Medicine, University of Oslo, Oslo, Norway and Department of Research, Vestre Viken, Drammen, Norway), Dr. Lars Ottestad, MD (Department of Genetics, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Oslo, Norway), Prof. Em. Rolf Kåresen, MD (Department of Oncology, Oslo University Hospital and Faculty of Medicine, University of Oslo, Oslo, Norway), Dr. Anita Langerød, PhD (Department of Genetics, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Oslo, Norway), Dr. Ellen Schlichting, MD (Department for Breast and Endocrine Surgery, Oslo University Hospital Ullevaal, Oslo, Norway), Dr. Marit Muri Holmen, MD (Department of Radiology and Nuclear Medicine, Oslo University Hospital, Oslo, Norway), Prof. Toril Sauer, MD (Department of Pathology at Akershus University hospital, Lørenskog, Norway), Dr. Vilde Haakensen, MD (Department of Genetics, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Oslo, Norway), Dr. Olav Engebråten, MD (Institute for Clinical Medicine, Faculty of Medicine, University of Oslo and Department of Oncology, Oslo University Hospital, Oslo, Norway), Prof. Bjørn Naume, MD (Division of Cancer Medicine and Radiotherapy, Department of Oncology, Oslo University Hospital Radiumhospitalet, Oslo, Norway), Dr. Cecile E. Kiserud, MD (National Advisory Unit on Late Effects after Cancer Treatment, Department of Oncology, Oslo University Hospital, Oslo, Norway and Department of Oncology, Oslo University Hospital, Oslo, Norway), Dr. Kristin V. Reinertsen, MD (National Advisory Unit on Late Effects after Cancer Treatment, Department of Oncology, Oslo University Hospital, Oslo, Norway and Department of Oncology, Oslo University Hospital, Oslo, Norway), Assoc. Prof. Åslaug Helland, MD (Department of Genetics, Institute for Cancer Research and Department of Oncology, Oslo University Hospital Radiumhospitalet, Oslo, Norway), Dr. Margit Riis, MD (Dept of Breast- and Endocrine Surgery, Oslo University Hospital, Ullevål, Oslo, Norway), Dr. Ida Bukholm, MD (Department of Breast-Endocrine Surgery, Akershus University Hospital, Oslo, Norway and Department of Oncology, Division of Cancer Medicine, Surgery and Transplantation, Oslo University Hospital, Oslo, Norway), Prof. Per Eystein Lønning, MD (Section of Oncology, Institute of Medicine, University of Bergen and Department of Oncology, Haukeland University Hospital, Bergen, Norway), Dr Silje Nord, PhD (Department of Genetics, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Oslo, Norway) and Grethe I. Grenaker Alnæs, M.Sc. (Department of Genetics, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Oslo, Norway). NBHS would like to thank study participants and research staff for their contributions and commitment to this study. OBCS thanks Meeri Otsukka and Kari Mononen. OFBCR thanks Teresa Selander and Nayana Weerasooriya. PBCS thanks Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, and Michael Stagner. SASBAC would like to thank the Swedish Medical Research Counsel. SBCS would like to thank Sue Higham, Helen Cramp, Ian Brock, Sabapathy Balasubramanian, and Dan Connley. SEARCH thanks the SEARCH and EPIC teams. SKKDKFZS thanks all study participants, clinicians, family doctors, researchers and technicians for their contributions and commitment to this study. TNBCC thanks Robert Pilarski and Charles Shapiro who were instrumental in the formation of the OSU Breast Cancer Tissue Bank, and also thanks the Human Genetics Sample Bank for processing of samples and providing OSU Columbus area control samples. UKBGS would like to thank Breast Cancer Now and the Institute of Cancer Research for support and funding of the Breakthrough Generations Study, and the study participants, study staff, and the doctors, nurses and other health care providers and health information sources who have contributed to the study, and acknowledge the NHS funding to the Royal Marsden/ICR NIHR Biomedical Research Centre. kConFab/AOCS wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and many families who contribute to kConFab. pKARMA would like to thank the Swedish Medical Research Counsel.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Financial supports
Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under grant agreement number 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (NIH, CA128978, CA122443) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. BCAC is funded by Cancer Research UK (C1287/A10118, C1287/A12014) and by the European Community´s Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2-2009-223175) (COGS). The ABCFS study was supported by grant UM1 CA164920 from the National Cancer Institute (USA). This study was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. The ABCS study was supported by the Dutch Cancer Society (grants NKI 2007-3839; 2009 4363), and Biobanking and BioMolecular resources Research Infrastructure—Netherlands (BBMRI-NL), which is a Research Infrastructure financed by the Dutch government (NWO 184.021.007). The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BBCS study was funded by Cancer Research UK and Breakthrough Breast Cancer and acknowledges National Health Service (NHS) funding to the National Institute for Health Research (NIHR) Biomedical Research Centre, and the National Cancer Research Network (NCRN). The BIGGS study was supported by NIHR Comprehensive Biomedical Research Centre, Guy’s & St. Thomas’ NHS Foundation Trust in partnership with King’s College London, United Kingdom. IT was supported by the Oxford Biomedical Research Centre. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the Deutsches Krebsforschungszentrum (DKFZ). The CECILE study was funded by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Ligue contre le Cancer Grand Ouest, Agence Nationale de Sécurité Sanitaire (ANSES), Agence Nationale de la Recherche (ANR). The CGPS study was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council and Herlev Hospital. The CNIO-BCS study was supported by the Instituto de Salud Carlos III, the Red Temática de Investigación Cooperativa en Cáncer and grants from the Asociación Española Contra el Cáncer and the Fondo de Investigación Sanitario (PI11/00923 and PI12/00070). The CTS study was initially supported by the California Breast Cancer Act of 1993 and the California Breast Cancer Research Fund (contract 97-10500) and is currently funded through the NIH (R01 CA77398). Collection of cancer incidence data (GLOBOCAN 2012) was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Sect. 103885. HAC received support from the Lon V Smith Foundation (LVS39420). The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). The GC-HBOC study was supported by the German Cancer Aid (grant no 110837, coordinator: Rita K. Schmutzler). The GENICA study was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, DKFZ, Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. The HEBCS study was financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (266528), the Finnish Cancer Society, the Nordic Cancer Union and the Sigrid Juselius Foundation. The HMBCS study was supported by a grant from the Friends of Hannover Medical School and by the Rudolf Bartling Foundation. The KBCP study was financially supported by the special Government Funding (EVO) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. The LMBC study was supported by the ‘Stichting tegen Kanker’ (232-2008 and 196-2010). The MARIE study was supported by the Deutsche Krebshilfe e.V. (70-2892-BR I, 106332, 108253, 108419), the Hamburg Cancer Society, DKFZ and the Federal Ministry of Education and Research (BMBF) Germany (01KH0402). The MBCSG study was supported by grants from the Italian Association for Cancer Research (AIRC) and by funds from the Italian citizens who allocated the 5/1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects “5 × 1000″). The MCBCS study was supported by the NIH grants CA128978, CA116167, CA176785 and NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), and the Breast Cancer Research Foundation and a generous gift from the David F. and Margaret T. Grohne Family Foundation and the Ting Tsung and Wei Fong Chao Foundation. The MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. This study was further supported by Australian NHMRC grants 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index. The MEC study was support by NIH grants CA63464, CA54281, CA098758 and CA132839. The work of MTLGEBCS was supported by the Quebec Breast Cancer Foundation, the Canadian Institutes of Health Research (CIHR) for the “CIHR Team in Familial Risks of Breast Cancer” program—grant # CRN-87521 and the Ministry of Economic Development, Innovation and Export Trade—grant # PSR-SIIRI-701. The NBCS study has received funding from the K.G. Jebsen Centre for Breast Cancer Research, the Research Council of Norway grant 193387/V50 (to A-L Børresen-Dale and V.N. Kristensen) and grant 193387/H10 (to A-L Børresen-Dale and V.N. Kristensen), South Eastern Norway Health Authority (grant 39346 to A-L Børresen-Dale) and the Norwegian Cancer Society (to A-L Børresen-Dale and V.N. Kristensen). The NBHS study was supported by NIH grant R01CA100374. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The OBCS study was supported by research grants from the Finnish Cancer Foundation, the Academy of Finland (grant number 250083, 122715 and Center of Excellence grant number 251314), the Finnish Cancer Foundation, the Sigrid Juselius Foundation, the University of Oulu, the University of Oulu Support Foundation and the special Governmental EVO funds for Oulu University Hospital-based research activities. The OFBCR study was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The PBCS study was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health and the Susan G. Komen Breast Cancer Foundation. The SBCS study was supported by Yorkshire Cancer Research S295, S299, S305PA and Sheffield Experimental Cancer Medicine Centre. The SEARCH study was funded by a programme grant from Cancer Research UK (C490/A10124) and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The SKKDKFZS study was supported by the DKFZ. The SZBCS study was supported by Polish State Committee for Scientific Research Grant PBZ_KBN_122/P05/2004. The TNBCC study was supported by: a Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a grant from the Breast Cancer Research Foundation, a generous gift from the David F. and Margaret T. Grohne Family Foundation, the Stefanie Spielman Breast Cancer fund and the OSU Comprehensive Cancer Center, the Hellenic Cooperative Oncology Group research grant (HR R_BG/04) and the Greek General Secretary for Research and Technology (GSRT) Program, Research Excellence II, the European Union (European Social Fund—ESF), and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF)—ARISTEIA. The UKBGS study was funded by Breast Cancer Now and the Institute of Cancer Research (ICR), London. ICR acknowledged NHS funding to the NIHR Biomedical Research Centre. The kConFab study was supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Financial support for the AOCS was provided by the United States Army Medical Research and Materiel Command (DAMD17-01-1-0729), Cancer Council Victoria, Queensland Cancer Fund, Cancer Council New South Wales, Cancer Council South Australia, the Cancer Foundation of Western Australia, Cancer Council Tasmania and the National Health and Medical Research Council of Australia (NHMRC; 400413, 400281, 199600). The pKARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer.
Additional information
Jieping Lei and Anja Rudolph share the first authorship.
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2015_1616_MOESM3_ESM.tif
ESM_3_QQPlot.tif Quantile–quantile plot for genotyped SNPs included in this analysis for associations with overall breast cancer risk (excluding SNPs located within TGFBR2 and CCND1)
439_2015_1616_MOESM4_ESM.pdf
ESM_4_Association_SNPs.pdf Associations with overall breast cancer risk for 3595 SNPs in the immunosuppression pathway genes
439_2015_1616_MOESM5_ESM.pdf
ESM_5_TopSNPs_ERstatus.pdf Associations of TGFBR2, CCND1 and STAT3 SNPs with overall breast cancer risk as well as stratified by ER status
439_2015_1616_MOESM7_ESM.tif
ESM_7_ForestPlot_rs1905339.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs1905339 with breast cancer risk
439_2015_1616_MOESM8_ESM.tif
ESM_8_ForestPlot_rs8074296.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs8074296 with breast cancer risk
439_2015_1616_MOESM9_ESM.tif
ESM_9_ForestPlot_rs146170568.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs146170568 with breast cancer risk
439_2015_1616_MOESM10_ESM.tif
ESM_10_ForestPlot_rs141732716.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs141732716 with breast cancer risk
439_2015_1616_MOESM11_ESM.tif
ESM_11_ForestPlot_rs138391971.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs138391971 with breast cancer risk
439_2015_1616_MOESM12_ESM.tif
ESM_12_ForestPlot_rs12952342.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs12952342 with breast cancer risk
439_2015_1616_MOESM13_ESM.tif
ESM_13_ForestPlot_rs190765034.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs190765034 with breast cancer risk
439_2015_1616_MOESM14_ESM.tif
ESM_14_ForestPlot_rs190137766.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs190137766 with breast cancer risk
439_2015_1616_MOESM15_ESM.tif
ESM_15_ForestPlot_chr17_40607850_I.tif Forest plot showing meta-analysis of study-wise estimates for the association of chr17:40607850:I with breast cancer risk
439_2015_1616_MOESM16_ESM.tif
ESM_16_ForestPlot_rs77942990.tif Forest plot showing meta-analysis of study-wise estimates for the association of rs77942990 with breast cancer risk
439_2015_1616_MOESM17_ESM.pdf
ESM_17_Gene_level_associations.pdf Gene-level associations with breast cancer risk for 133 candidate genes in the immunosuppression pathway
439_2015_1616_MOESM20_ESM.tif
ESM_20_Boxplot_STAT3.tif Box plot showing gene expression levels of STAT3 in normal breast tissue as well as tumor breast tissue
439_2015_1616_MOESM21_ESM.tif
ESM_21_Boxplot_IL5.tif Box plot showing gene expression levels of IL5 in normal breast tissue as well as tumor breast tissue
439_2015_1616_MOESM22_ESM.tif
ESM_22_Boxplot_PTRF.tif Box plot showing gene expression levels of PTRF in normal breast tissue as well as tumor breast tissue
439_2015_1616_MOESM23_ESM.tif
ESM_23_Boxplot_CSF2.tif Box plot showing gene expression levels of GM-CSF in normal breast tissue as well as tumor breast tissue
439_2015_1616_MOESM24_ESM.tif
ESM_24_eQTL_rs1905339_STAT3.tif Associations of rs1905339 genotypes with STAT3 expression within 183 breast tissue samples
439_2015_1616_MOESM25_ESM.tif
ESM_25_eQTL_rs8074296_STAT3.tif Associations of rs8074296 genotypes with STAT3 expression within 183 breast tissue samples
439_2015_1616_MOESM26_ESM.tif
ESM_26_eQTL_rs77942990_STAT3.tif Associations of rs8074296 genotypes with STAT3 expression within 183 breast tissue samples
439_2015_1616_MOESM27_ESM.tif
ESM_27_eQTL_rs1905339_PTRF.tif Associations of rs1905339 genotypes with PTRF expression within 183 breast tissue samples
439_2015_1616_MOESM28_ESM.tif
ESM_28_eQTL_rs8074296_PTRF.tif Associations of rs8074296 genotypes with PTRF expression within 183 breast tissue samples
439_2015_1616_MOESM29_ESM.tif
ESM_29_eQTL_rs77942990_PTRF.tif Associations of rs77942990 genotypes with PTRF expression within 183 breast tissue samples
439_2015_1616_MOESM30_ESM.tif
ESM_30_eQTL_rs1905339_TUBG2.tif Associations of rs1905339 genotypes with TUBG2 expression within 183 breast tissue samples
439_2015_1616_MOESM31_ESM.tif
ESM_31_eQTL_rs8074296_ TUBG2.tif Associations of rs8074296 genotypes with TUBG2 expression within 183 breast tissue samples
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lei, J., Rudolph, A., Moysich, K.B. et al. Genetic variation in the immunosuppression pathway genes and breast cancer susceptibility: a pooled analysis of 42,510 cases and 40,577 controls from the Breast Cancer Association Consortium. Hum Genet 135, 137–154 (2016). https://doi.org/10.1007/s00439-015-1616-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-015-1616-8