
Abstract
To enhance the marker density in the “QTL-hotspot” region, harboring several QTLs for drought tolerance-related traits identified on linkage group 04 (CaLG04) in chickpea recombinant inbred line (RIL) mapping population ICC 4958 × ICC 1882, a genotyping-by-sequencing approach was adopted. In total, 6.24 Gb data from ICC 4958, 5.65 Gb data from ICC 1882 and 59.03 Gb data from RILs were generated, which identified 828 novel single-nucleotide polymorphisms (SNPs) for genetic mapping. Together with these new markers, a high-density intra-specific genetic map was developed that comprised 1,007 marker loci spanning a distance of 727.29 cM. QTL analysis using the extended genetic map along with precise phenotyping data for 20 traits collected over one to seven seasons identified 49 SNP markers in the “QTL-hotspot” region. These efforts have refined the “QTL-hotspot” region to 14 cM. In total, 164 main-effect QTLs including 24 novel QTLs were identified. In addition, 49 SNPs integrated in the “QTL-hotspot” region were converted into cleaved amplified polymorphic sequence (CAPS) and derived CAPS (dCAPS) markers which can be used in marker-assisted breeding.
Similar content being viewed by others


Introduction
Chickpea (Cicer arietinum L.) is the world’s second most important food legume crop, cultivated primarily on marginal lands in the arid and semi-arid regions of South Asia and sub-Saharan Africa. It is a self-pollinated species with basic chromosome number eight and genome size of 738 Mb (Varshney et al. 2013a). Globally, chickpea is cultivated on 13.5 Mha with an annual production of 13.1 Mt (FAO 2013) and is a rich source of protein especially in vegetarian diets. However, chickpea production is affected by various biotic stresses including Fusarium wilt, Ascochyta blight, Helicoverpa and abiotic stresses such as drought, heat and salinity. As chickpea is predominantly cultivated on residual soil moisture, terminal drought is a serious problem and will become more prevalent due to climate change and global warming (Tuberosa 2012; Dodig et al. 2012).
During the past three decades, there has been a shift in cultivation of chickpea from cooler to warmer regions both in Asia and Africa (Kimurto et al. 2014; Krishnamurthy et al. 2013) which has also increased the impact of drought on productivity. Therefore, enhancing the drought tolerance in chickpea would help to stabilize and increase production. Marker-assisted selection (MAS) has already proved its importance in accelerating the process of variety development (Varshney et al. 2005). However, breeding efforts towards developing drought-tolerant chickpea varieties have remained slow, mainly because of precision issues in phenotyping for drought tolerance, narrow genetic base and the limited availability of genomic resources. Nevertheless, in recent years, the availability of large-scale genomic resources (Varshney et al. 2009; Nayak et al. 2010; Gujaria et al. 2011; Thudi et al. 2011; Hiremath et al. 2012) and high throughput phenotyping (Kashiwagi et al. 2013) have facilitated progress towards the genetic analysis of drought tolerance in chickpea. With the increasing efforts, QTLs for drought-related traits have been identified in several studies (Rehman et al. 2011; Hamwieh et al. 2013; Jamalabadi et al. 2013), though their validation has not yet been reported. Recently, Varshney et al. (2014a) reported 45 robust main-effect QTLs (M-QTLs; QTLs which explain >10 % phenotypic variation (PVE) and 973 epistatic QTLs (E-QTLs; explaining 58.2 and 92.19 % PVE), respectively, using two intra-specific RIL mapping populations (ICC 4958 × ICC 1882 and ICC 283 × ICC 8261). In addition, the study also revealed nine QTL clusters including a genomic region on CaLG04 referred to as “QTL-hotspot”, harboring several QTLs for drought tolerance-related traits. Introgression of this “QTL-hotspot” in one elite variety, JG 11, has shown improvement of drought tolerance-related traits (Varshney et al. 2013b). However, the “QTL-hotspot” was genetically large (~29 cM on the genetic map and 7.74 Mb on the physical map; Varshney et al. 2014b) and was associated with relatively few SSR markers making it difficult to identify polymorphism between the recurrent and donor genotypes in a backcrossing strategy and also to identify genes associated with drought tolerance in this region (Thudi et al. 2014). Enriching this region with additional markers will facilitate fine mapping and precision breeding for drought tolerance.
Single-nucleotide polymorphism (SNP) markers have become the markers of choice due to their high abundance and cost efficiency, primarily due to advances in sequencing technologies and their application to genotyping crop species (Silvar et al. 2011). For instance, the genotyping-by-sequencing (GBS) approach proposed by Elshire et al. (2011) has increased the efficiency of SNP discovery and genotyping by enabling high multiplexing of samples and simple library preparation procedures. This approach is now being used in several crops for diversity assessment, trait mapping, genome-wide association studies and genomic selection (Deschamps et al. 2012; Poland and Rife 2012).
In this study, the GBS approach was used to identify and genotype SNPs in an intra-specific mapping population ICC 4958 × ICC 1882 in which the “QTL-hotspot” region was identified. As a result, several novel SNPs were integrated into “QTL-hotspot” region and converted to cleaved amplified polymorphic sequences (CAPS) and derived CAPS (dCAPS) markers that can be used cost effectively for molecular breeding to improve drought tolerance in chickpea.
Materials and methods
Plant material
One intra-specific recombinant inbred line (RIL) mapping population derived from ICC 4958 × ICC 1882 (ICCRIL03) comprising of 264 individuals was used in the present study. Detailed account on parental lines and mapping population are provided in Varshney et al. (2014a). DNA was isolated from 232 RILs and parental genotypes using high throughput mini-DNA extraction method as described by Cuc et al. (2008). The quality and quantity of DNA were checked using spectrophotometer (Shimadzu UV160A, Japan). Two hundred and eight RILs with high-quality DNA were selected for sequencing.
Genotyping-by-sequencing (GBS)
A GBS approach was used for SNP calling between the parents and genotyping the RILs as described by Elshire et al. (2011). In brief, the GBS libraries from the parental lines and RILs were prepared using ApeKI endonuclease (recognition site: G/CWCG) and sequenced using the Illumina HiSeq 2000 platform (Illumina Inc, San Diego, CA, USA). Genomic DNA of selected mapping population and parental lines were subjected for restriction digestion using endonuclease ApeKI for 2 h at 75 °C. Adapters with unique multiplex sequence index (barcodes) were ligated to the sticky ends using ligase buffer with ATP and T4 ligase. Samples were incubated at 22 °C for 1 h and heated to 65 °C for 30 min to inactivate the T4 ligase. Aliquot of each sample (5 µl) was pooled (multiplexed) and purified to remove the excess adapters. DNA samples were eluted in a final volume of 50 µl. PCR was performed to increase the restriction fragments from each library using primers complementary to the corresponding adapters. The amplified pools constituting the “sequencing library,” were cleaned up and evaluated for fragment sizes using a DNA analyzer. Libraries without adapter dimers were subjected to sequencing.
SNP calling
The reads obtained were first de-multiplexed according to the sample barcodes and adapter sequences were removed using custom perl script. The reads having more than 50 % of low quality base pairs (Phred <5 %) were discarded and filtered data were used for calling SNPs after quality check (Q score >20). The filtered, high-quality data from each sample was aligned to the draft genome sequence (CaGAv1.0) of chickpea (Varshney et al. 2013a) using SOAP (Li et al. 2009). The nucleotide with highest probability at each position under a Bayesian model was identified for individual RILs and the consensus sequences were saved in FASTA format. Consensus sequences from all samples were compared to detect polymorphic loci. Polymorphic loci that were either heterozygous in any of the parents or present in <50 % individuals in the population were discarded and a high-quality SNP dataset was generated.
Linkage mapping
Genotyping data generated in this and previous studies (See Online Resource 1) were compiled for linkage analysis using JoinMap V4.0 (Van Ooijen and Voorrips 2006). Marker order was assigned using the regression mapping algorithm with maximum recombination frequency of 0.4 at minimum logarithm of odds (LOD) of 3 and jump threshold of 5. Ripple command was used after adding each marker locus to confirm marker order. The Kosambi mapping function was used to calculate the map distance (Kosambi 1943). To detect segregation distortion, Chi-square (χ 2) values were calculated using Joinmap V4.0. Highly distorted and unlinked markers were excluded from analysis. Mapchart 2.2 (Voorrips 2002) was used to visualize a constructed map for each linkage group. Linkage groups were named according to Varshney et al. (2014a).
QTL analysis
Genotyping data obtained in the current study and the phenotyping data for 20 drought tolerance-related traits including root traits, morphological, phenological, yield, yield-related traits and drought indices (as mentioned in Varshney et al. 2014a) were used for QTL analysis using QTL Cartographer V.2.5 software (Wang et al. 2012). Composite interval mapping (CIM) was performed by selecting Model 6 with the default window size 10 cM, control marker number 5, and backward regression method. To obtain more precise results the default walk speed was reduced to 1 cM. LOD method (LOD > 3) was used to determine the significance of each QTL interval with the threshold level performed at 1,000 permutations, significance level of p ≤ 0.05.
Conversion of SNPs into CAPS and dCAPS
Single-nucleotide polymorphisms (SNPs) integrated in the “QTL-hotspot” region were converted to CAPS and dCAPS using dCAPS Finder 2.0 (Neff et al. 2002). The predicted CAPS and dCAPS candidates were amplified in a 20 µl PCR reaction using GeneAmp® PCR System 9700 thermal cycler (Applied Biosystems, Foster City, CA, USA) on 5 parental genotypes of chickpea inter and intra-specific mapping populations (PI 489777, ICC 4958, ICC 1882, ICC 8261 and ICC 283). Amplicons for each CAPS and dCAPS were subjected to digestion using their respective restriction enzymes followed by separation on 2 % agarose gel electrophoresis as described in Gujaria et al. (2011). Details about these primer sequences, PCR conditions and product size are given in Online Resource 2.
Identification of candidate genes
The amino acid sequences predicted from gene models of genes located in the region delimited by the “QTL-hotspot” were retrieved from draft genome sequence (CaGAv1.0) of chickpea (Varshney et al. 2013a; http://www.icrisat.org/gt-bt/ICGGC/GenomeManuscript.htm) and searched against NCBI-nr protein database using blast program implemented in Blast2GO software (Conesa et al. 2005) with an E value threshold of ≤e−20. Associated gene ontology (GO) terms were exported and searched for plant-related GO terms using the GO slim viewer from the AgBase web server (http://www.agbase.msstate.edu), which also categorize terms into three different classes as biological processes (BP), molecular function (MF) and cellular components (CC).
Results
Sequence data and SNP discovery
Parental genotypes of the mapping population (ICC 4958 × ICC 1882) were sequenced at higher depth (5× coverage) than RIL individuals, and a total of 69.39 million reads containing 6.24 Gb for ICC 4958 and 62.79 million reads containing 5.65 Gb for ICC 1882 were generated. In addition, 701.05 million reads containing 59.03 Gb were generated for 208 RILs. The number of reads generated varied from 0.28 million (RIL078) to 19.23 million (RIL204) with an average of 3.37 million per line. The data obtained were filtered and used for SNP identification using SOAP software. The SNPs identified were again parsed to remove heterozygous SNPs in parents and a set of 828 SNPs were identified across 208 RILs. The flanking sequences of all SNPs have been provided in Online Resource 3.
Construction of genetic map
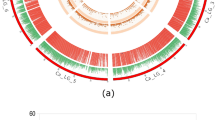
Genotypic data for 828 polymorphic SNPs generated in this study along with 318 markers (including 241 markers from Varshney et al. 2014a) obtained from the earlier studies (Online Resource 1) were used for genetic map construction. In total, 1,146 markers were used for genetic map construction, of which 1,007 (87.87 %) could be mapped on eight linkage groups (CaLG01–CaLG08) covering 727.29 cM (Fig. 1; http://cmap.icrisat.ac.in/cmap/sm/cp/jaganathan/). These included 743 SNPs, 232 simple sequence repeats (SSRs), 21 diversity arrays technology (DArT), 7 expressed sequence tag-SSR (EST-SSR) and 4 genic molecular markers (GMM) (Table 1). The highest number of markers was mapped on CaLG04 (386), while the lowest number of markers was mapped to CaLG05 (39). The distribution of marker loci on 8 linkage groups has been shown in Fig. 1 and Online Resource 4.



High-density intra-specific genetic map of chickpea (ICC 4958 × ICC 1882). This map is comprised of 1,007 markers including 743 novel SNPs from GBS approach and spans 727.29 cM. Genetic distances (cM) were shown on the left side and the markers were shown on the right side of the bars. Map was constructed using JoinMap 4.0 and Kosambi function. Markers in black color font are from the framework map and markers in red color font are newly generated SNP markers. For clear visualization, the CaLG04 and CaLG06 were split into two parts and named as A, B
The length of the linkage groups varied from 59.41 cM (CaLG05) to 112.10 cM (CaLG04). The highest marker density was observed for CaLG04, which had 3.44 markers per cM on average, whereas lowest marker density was observed for CaLG07, which had 0.62 markers per cM on average. Overall, the map had 1.30 markers per cM on average (Table 1; Fig. 1). Of 828 SNPs used for linkage map construction, 743 SNPs (89.73 %) were mapped, among which 342 were mapped on CaLG04. Of 279 SSR markers used, 232 (83.15 %) were mapped. Comparatively, SSR markers were mapped evenly on all the eight linkage groups, the highest number of SSR markers was mapped on CaLG03 (47), and the lowest number of SSR markers was mapped on CaLG02 (18). Out of 14 EST-SSRs, 50 % were mapped, whereas all GMM and DArT markers used in the present study were mapped. However, among 21 DArT markers mapped, 47.6 % (10) were on CaLG01, 33.33 % (7) on CaLG03, one each on CaLG02, CaLG05, CaLG06 and CaLG07. Out of four GMM markers, one each was mapped on CaLG02, CaLG04, CaLG07 and CaLG08.
Marker enrichment in the “QTL-hotspot” region
QTL analysis based on genotypic data for 1,007 markers and phenotypic data for 20 traits (as described in Varshney et al. 2014a), identified a total of 164 robust main-effect QTLs (M-QTLs) by QTL Cartographer 2.5. More than 50 % (91 M-QTLs) of these M-QTLs were located on CaLG04 and significantly, all 91 QTLs were detected in the “QTL-hotspot” region (Online Resource 5). The earlier reported “QTL-hotspot” region (Varshney et al. 2014a) had 7 SSR markers (ICCM0249, NCPGR127, TAA170, NCPGR21, TR11, GA24 and STMS11) and spanned 29 cM on linkage group CaLG04. The current study integrates 49 new SNP markers in the “QTL-hotspot” region spanning 14 cM (Fig. 2).

Saturated “QTL-hotspot” region with additional markers. The figure shows comparison of the “QTL-hotspot” updated with 49 novel SNP markers in this study and with the one reported by Varshney et al. (2014a)
QTL analysis
Out of 20 traits analyzed, QTLs were identified for 16, including root length density (RLD, cm cm−3), root surface area (RSA, cm2), root dry weight/total plant dry weight (RTR, %), shoot dry weight (SDW, g), plant height (PHT, cm), primary branches (PBS), days to 50 % flowering (DF), days to maturity (DM), 100-seed weight (100 SDW, g), biomass (BM, g), harvest index (HI, %), pods/plant (POD), seeds/pod (SPD), yield (YLD, g), drought susceptibility index (DSI) and drought tolerance index (DTI). QTL nomenclature was adopted as per Varshney et al. (2014a). In case a QTL reported for a given trait in (Varshney et al. 2014a) has been further resolved into two or more QTLs, the QTLs are further named using a decimal followed by a roman numeral. For instance QR3rld01 reported earlier was resolved into three, hence named as QR3rld01.1, QR3rld01.2, QR3rld01.3 (Online Resource 5). QTLs were considered as ‘stable’ (if they appeared in more than one location for the specified trait) and ‘consistent’ (if they appear in more than 1 year/season for the specified trait) as described in Varshney et al. (2014a). Identified QTLs are discussed below.
Root-related traits
Three QTLs were identified, one each for RLD, RSA and RTR with PVE ranging from 10.65 to 13.56 % (Table 2). Among them, RLD and RTR were identified in the “QTL-hotspot” as reported earlier, whereas a QTL for RSA was identified on CaLG06 (Online Resource 5). The QTL for RLD, ‘QR3rld01’ was refined to 3.23–5.37 cM from 10.54 cM, whereas that for RTR, ‘QR3rtr01’ was refined to 1.81–5.37 cM from 5 cM (Table 2, Online Resource 5). Both QTLs were consistent across years (2005 and 2007).
Morphology-related traits
A total of 3 and 9 QTLs were identified for SDW and PHT, respectively, out of which 2 and 5 were newly identified for the respective traits. Overall, PVE ranged from 10.05 to 34.57 % (Table 2). The QTL size for SDW ‘QR3sdw01’ was similar as reported earlier, whereas QTL for PHT, ‘QR3pht03’ was refined to 1.81 cM from 5.37 cM (Online Resource 5). Out of 9 QTLs identified for PHT, 3 were stable and 5 were consistent. Interestingly, two QTLs, ‘QR3pht06’, and ‘QR3pht08’ were found consistent and stable, which were previously reported to be unstable and inconsistent by Varshney et al. (2014a). A QTL for primary branches (PBS) ‘QR3pbs02’ was newly identified in the current study which explained the PVE of 12.92 % (Table 2 and Online Resource 5).
Phenology-related traits
For phenological traits, 3 and 2 QTLs were identified for DF and DM, respectively. The maximum phenotypic variation explained by the QTL, ‘QR3df04’ was much higher (67.71 %) as compared to the earlier study (26.87 %) for DF (Online Resource 5). This QTL has been refined to 1.81 cM from 5.14 cM. Similarly, the QTL, ‘QR3dm01’ explained 47.43 % PVE for DM which was comparatively higher than that reported earlier (19.71 %) and was refined to 7.33 cM from 15.13 cM (Table 2 and Online Resource 5).
Yield-related traits and drought indices
A total of 16 QTLs including 5 novel QTLs were identified for yield and yield-related traits, including 2 each for 100 SDW and POD and 3 each for BM, HI, SPD and YLD. Overall, the QTLs spanned the same size as reported previously (Varshney et al. 2014a); however, QTLs for BM and POD have been refined to 15.13 and 5.37 cM, respectively (Table 2). The PVE by each QTL was comparatively high, especially for 100SDW, ‘QR3100sdw03,’ which had PVE of 60.41 % (Online Resource 5).
In the case of drought indices, a novel QTL, ‘QR3dsi02’, was identified explaining 13.00 % phenotypic variation for DSI, whereas no QTL for DSI was reported in the earlier study (Varshney et al. 2014a) (Table 2). Three QTLs were identified for DTI, of which 2 were novel. Interestingly, a QTL from the “QTL-hotspot”, ‘QR3dti02’, which was earlier reported to be a minor QTL, was ranked as robust in this study (Table 2 and Online Resource 5).
CAPS and dCAPS marker assays
As breeders are interested in an inexpensive and technically less demanding genotyping platform for marker-assisted breeding, the SNPs integrated into the “QTL-hotspot” were converted to CAPS/dCAPS markers. A total of 16 CAPS and 33 dCAPS primer pairs were designed and verified for amplification (Online Resource 2). However, only 20 out of 49 primer pairs showed single prominent amplicon and subsequently used for restriction digestion on a panel of 5 parental genotypes. As a result, 14 CAPS and 1 dCAPS were developed (Online Resource 6). In total, 8 CAPS markers were polymorphic in inter-specific mapping population PI 489777 × ICC 4958, while 14 (13 CAPS and 1 dCAPS) each in two intra-specific mapping populations ICC 4958 × ICC 1882 and ICC 283 × ICC 8261 were polymorphic (Online Resource 2).
Selection of candidate genes
A detailed analysis of QTLs from the “QTL-hotspot” region showed that, QTLs for 9 traits (RTR, SDW, PHT, DF, 100SDW, DM, HI, SPD and DTI) were flanked by Ca4_11276225 and Ca4_12558541 markers. Further, the traits RTR, RLD, PHT, DF, DM, 100 SDW, BM, POD and YLD were flanked by Ca4_13687456 and NCPGR21 markers. As the QTLs for 13 out of 16 traits fall between markers Ca4_11276225 and NCPGR 21 (whose physical position on genome is 14,146,315 bp), the ~3 Mb region between these markers was selected for candidate gene identification (Online Resource 5). The 3 Mb region contained 286 genes. The amino acid sequences for these 286 genes were searched against the NCBI-nr protein database. Of these, 211 sequences were annotated and 1,050 GO terms were obtained (Online Resource 7 and 8). Categorization of these terms into BP, MF and CC showed predominance of stress-related GO terms in BP class, while in MF class GO terms for binding, catalytic, transferase, hydrolyses and kinase activity were predominantly present. Genes having a direct role in stress such as dehydration-responsive element-binding protein (DREB), heat stress transcription protein, thiamine thiazole synthase and few uncharacterized proteins were also identified in the region (Online Resource 7 and 8).
Discussion
Drought seems to continue to be a serious constraint to chickpea production. Owing to its complex nature, the genetic dissection of drought tolerance into component traits has been challenging. However, comprehensive insights have been provided into component traits by Varshney et al. (2014a). The reported “QTL-hotspot” required mapping refinement to allow QTL cloning for component trait improvement through molecular breeding. In the present study, efforts were made to saturate this region to facilitate fine mapping.
SNP markers and linkage mapping
To date, primarily SSR markers have been used for linkage mapping in chickpea intra-specific populations. Although availability of genomic resources has reduced the SSR marker identification span, polymorphism study and further screening is still a time-consuming and labor-intensive process. As a result, most genetic maps remain limited to only a few hundred markers (Radhika et al. 2007; Jamalabadi et al. 2013; Varshney et al. 2014a). We used a GBS approach which has the advantage of simultaneous SNP identification and genotyping. As a result, we identified 828 novel SNPs. Thus, a greater number of markers are now available for this intra-specific population. As compared to GBS studies in other plant species, SNP markers identified in the present study were less (Poland et al. 2012; Sonah et al. 2013). This might be because of variable number of reads generated per RIL (0.28–19.23 million reads) resulting in more missing data points or very stringent SNP calling criterion adopted, for instance SNPs present in <50 % RILs were excluded.
A total of 1,146 markers were used for linkage map construction, out of which 1,007 (87.87 %) markers were mapped which spanned 727.29 cM. This saturated map has approximatively fourfold more markers and increases the marker density from 0.50 to 1.30 per cM as compared to the previous 241 loci map (Varshney et al. 2014a). Nearly, 94.60 % (228) markers from the earlier study (Varshney et al. 2014a), were mapped on the respective linkage groups in the new map, reflecting the higher level of conservation in marker order between the maps. Interestingly, 46 % of the SNP markers were mapped on CaLG04. This may be due to high repeat-rich regions in the case of Ca4 pseudomolecule and it was evident from our earlier studies (Varshney et al. 2013a), that the average SNP density per Kb (7.6) is higher in the case of chickpea “Ca4” psuedomolecule, i.e., the CaLG04. Further, the study also indicated higher diversity level in elite cultivars of chickpea in the case of “Ca4” pseudomolecule (Theta Pi = 2.8180; Theta w = 2.2377). High Theta π and Theta w are usually associated with repeat-rich regions in genome.
Refining the “QTL-hotspot” and developing breeder-friendly markers
The current analysis integrated 49 new SNP markers in the “QTL-hotspot” region thereby enriching the same from 7 markers to 55 markers (among 7 previously mapped SSRs, two SSR markers GA24 and TR11 could not be mapped; however, ICCM0065 was newly mapped in this region). Integration of these 49 markers has refined the “QTL-hotspot” region from 29 to 14 cM. Several fine mapping studies earlier have shown that the integration of additional markers has narrowed down the QTL interval. For instance, in the case of rice, Yu et al. (2011) demonstrated that mapping of additional SNP markers not only detected new QTLs but also increased the resolution of the QTLs. Similarly, Silvar et al. (2012) fine mapped the QTLs for powdery mildew resistance by integrating 32 markers in the QTL region in Spanish barley. Likewise, in case of basmati rice, the “aro3-1” QTL was narrowed down to an interval of 390 kb from the earlier reported interval of 8.6 Mb and “aro8-1” QTL was narrowed down to a physical interval of 430 kb (Singh et al. 2007).
The QTL analysis was performed for 20 different traits and 164 robust M-QTLs were detected for 16 traits which included all 14 reported traits from Varshney et al. (2014a). More than 50 % (91) of QTLs were located on CaLG04 and all were detected in the “QTL-hotspot” region which highlights the importance of this region in drought tolerance mechanism in chickpea. In addition, the current study also identified new QTLs for PBS and DSI which were not detected/reported earlier. Furthermore, some QTLs which were unstable, inconsistent in the earlier study (Varshney et al. 2014a) were identified to be stable and consistent. For instance, five additional QTLs were identified in the case of PHT and one additional QTL each for SDW, DF, BM, POD, SPD and yield (Online Resource 5). Comparatively, the PVE observed for most of the traits was significantly high, indicating robustness of the identified QTLs.
To enhance molecular breeding for introgressing the “QTL-hotspot”, SNP markers were converted into CAPS/dCAPS. As the SSR markers from the “QTL-hotspot” showed less/no polymorphism between ICC 4958 and few recurrent chickpea elite cultivars (Thudi et al. 2014), these CAPS and dCAPS markers would be of interest to breeders in marker-assisted breeding programs to introgress the “QTL-hotspot” region.
Candidate gene identification
Functional annotation of the candidate genes revealed their role in various abiotic and biotic stress tolerance mechanisms. For instance, dehydration-responsive element-binding protein (DREB) which is a well-known transcription factor involved in abiotic stress including drought tolerance (Liu et al. 1998; Lata and Prasad 2011) was identified in the “QTL-hotspot” region. Similarly, thiamine thiazole synthase, which was reported to be involved in stress-related mechanisms (Rapala-Kozik et al. 2012) was also identified in the “QTL-hotspot” region. In addition to these, few trait-specific genes like E3 ubiquitin–protein ligase and TIME FOR COFFE (TIC) were also identified. The E3 ubiquitin–protein ligase activity has been reported to be involved in grain width and weight in rice (Song et al. 2007) while TIC protein has been reported to play role in plant growth, development and circadian clock (Hall et al. 2003; Sanchez et al. 2011; Shin et al. 2013). Shin et al. (2012) has reported a role of TIC in jasmonic acid signaling pathways and in the control of root meristem size in Arabidopsis. Loss of this gene was reported to result in reduced root meristem length and cell number (Hong et al. 2014). Therefore, further fine mapping and cloning of genes underlying QTL would unravel the genetics behind drought tolerance in chickpea.
In summary, we implemented GBS approach for developing a high-density linkage map from an intra-specific population in chickpea. The map contains 1,007 loci spanning 727.29 cM and enriching the “QTL-hotspot” region from 7 markers to 55 markers. Also this study has refined the “QTL-hotspot” region from 29 to 14 cM on a genetic map corresponding to ~4 Mb on the physical map. The current study also identified the presence of several stress-related candidate genes including DREB in the "QTL-hotspot" region. Further characterization of these genes will help in identifying the mechanisms of drought tolerance in chickpea. In addition, the CAPS/dCAPS markers developed in this study can be used in marker-assisted breeding program for introgressing the “QTL-hotspot” into elite cultivar. Further study and marker enrichment of this region will facilitate fine mapping, QTL cloning and help in understanding the mechanism of drought tolerance in chickpea.
References
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Cuc LM, Mace ES, Crouch JH, Quang VD, Long TD, Varshney RK (2008) Isolation and characterization of novel microsatellite markers and their application for diversity assessment in cultivated groundnut (Arachis hypogaea). BMC Plant Biol 8:55
Deschamps S, Nannapaneni K, Zhang Y, Hayes K (2012) Local assemblies of paired end reduced representation libraries sequenced with the Illumina genome analyser in Maize. Int J Plant Genomics 2012:8
Dodig D, Zorić M, Kandić V, Perović D, Šurlan-Momirović G (2012) Comparison of responses to drought stress of 100 wheat accessions and landraces to identify opportunities for improving wheat drought resistance. Plant Breed 131:369–379
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6(5):e19379
FAOSTAT (2013) FAOSTAT database. http://faostat3.fao.org/faostat-gateway/go/to/download/Q/QC/E [Verified 02 September 2014]
Gujaria N, Kumar A, Dauthal P, Dubey A, Hiremath P, Prakash AB, Farmer A, Bhide M, Shah T, Gaur PM, Upadhyaya HD, Bhatia S, Cook DR, May GD, Varshney RK (2011) Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.). Theor Appl Genet 122:1577–1589
Hall A, Bastow RM, Davis SJ, Hanano S, McWatters HG, Hibberd V, Doyle MR, Sung SB, Halliday KJ, Amasino RM, Millar AJ (2003) The TIME FOR COFFEE gene maintains the amplitude and timing of Arabidopsis circadian clocks. Plant Cell 15:2719–2729
Hamwieh A, Imtiaz M, Malhotra RS (2013) Multi-environment QTL analyses for drought-related traits in a recombinant inbred population of chickpea (Cicer arietinum L.). Theor Appl Genet 126:1025–1038
Hiremath PJ, Kumar A, Penmetsa RV, Farmer A, Schlueter JA, Chamarthi SK, Whaley AM, Carrasquilla-Garcia N, Gaur PM, Upadhyaya HD, Kishor PBK, Shah TM, Cook DR, Varshney RK (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10:716–732
Hong LW, Yan DW, Liu WC, Chen HG, Lu YT (2014) TIME FOR COFFEE controls root meristem size by changes in auxin accumulation in Arabidopsis. J Exp Bot 65:275–286
Jamalabadi JG, Saidi A, Karami E, Kharkesh M, Talebi R (2013) Molecular mapping and characterization of genes governing time to flowering, seed weight, and plant height in an intraspecific genetic linkage map of chickpea (Cicer arietinum). Biochem Genet 51:387–397
Kashiwagi J, Krishnamurthy L, Gaur PM, Upadhyaya HD, Varshney RK, Tobita S (2013) Traits of relevance to improve yield under terminal drought stress in chickpea (C. arietinum L.). Field Crop Res 145:88–95
Kimurto PK, Mulwa RMS, Towett BK, Cheruiyot EK, Gangarao R, Silim S, Rutto DK, Kirui G, Gaur PM, Varshney RK (2014) Screening for drought and pod borer (Helicoverpa armigera) tolerance in selected chickpea (Cicer arietinum L.) germplasm in semi-arid areas of Kenya. Egerton J Sci Technol 9:23–30
Kosambi DD (1943) The estimation of map distances from recombination values. Ann Hum Genet 12:172–175
Krishnamurthy L, Kashiwagi J, Upadhyaya HD, Gowda CLL, Gaur PM, Singh S, Purushothaman R, Varshney RK (2013) Partitioning coefficient-a trait that contributes to drought tolerance in chickpea. Field Crop Res 149:354–365
Lata C, Prasad M (2011) Role of DREBs in regulation of abiotic stress responses in plants. J Exp Bot 62:4731–4748
Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Liu Q, Kasuga M, Sakuma Y, Abe H, Miura S, Yamaguchi-Shinozaki K, Shinozaki K (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought- and low-temperature-responsive gene expression, respectively, in Arabidopsis. Plant Cell 10:1391–1406
Nayak SN, Zhu HY, Varghese N, Datta S, Choi HK, Horres R, Jungling R, Singh J, Kishor PBK, Sivaramakrishnan S, Hoisington DA, Kahl G, Winter P, Cook DR, Varshney RK (2010) Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120:1415–1441
Neff MM, Turk E, Kalishman M (2002) Web-based primer design for single nucleotide polymorphism analysis. Trends Genet 18:613–615
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 5:92–102
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7(2):e32253
Radhika P, Gowda SJM, Kadoo NY, Mhase LB, Jamadagni BM, Sainani MN, Chandra S, Gupta VS (2007) Development of an integrated intraspecific map of chickpea (Cicer arietinum L.) using two recombinant inbred line populations. Theor Appl Genet 115:209–216
Rapala-Kozik M, Wolak N, Kujda M, Banas AK (2012) The upregulation of thiamine (vitamin B-1) biosynthesis in Arabidopsis thaliana seedlings under salt and osmotic stress conditions is mediated by abscisic acid at the early stages of this stress response. BMC Plant Biol 12:2
Rehman AU, Malhotra RS, Bett K, Tar’an B, Bueckert R, Warkentin TD (2011) Mapping QTL associated with traits affecting grain yield in chickpea (Cicer arietinum L.) under terminal drought stress. Crop Sci 51:450–463
Sanchez A, Shin J, Davis SJ (2011) Abiotic stress and the plant circadian clock. Plant Signal Behav 6:223–231
Shin J, Heidrich K, Sanchez-Villarreal A, Parker JE, Davis SJ (2012) TIME FOR COFFEE represses accumulation of the MYC2 transcription factor to provide time-of-day regulation of jasmonate signaling in Arabidopsis. Plant Cell 24:2470–2482
Shin J, Du SX, Bujdoso N, Hu YJ, Davis SJ (2013) Overexpression and loss-of-function at TIME FOR COFFEE results in similar phenotypes in diverse growth and physiological responses. J Plant Biol 56:152–159
Silvar C, Perovic D, Casas AM, Igartua E, Ordon F (2011) Development of a cost-effective pyrosequencing approach for SNP genotyping in barley. Plant Breed 130:394–397
Silvar C, Perovic D, Scholz U, Casas AM, Igartua E, Ordon F (2012) Fine mapping and comparative genomics integration of two quantitative trait loci controlling resistance to powdery mildew in a Spanish barley landrace. Theor Appl Genet 124:49–62
Singh R, Singh AK, Sharma TR, Singh A, Singh NK (2007) Fine mapping of aroma QTLs in basmati rice (Oryza sativa L.) on Chromosomes 3, 4 and 8. J Plant Biochem Biotechnol 16:75–82
Sonah H, Bastien M, Iquira E, Tardivel A, Legare G, Boyle B, Normandeau E, Laroche J, Larose S, Jean M, Belzile F (2013) An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS One 8(1):e54603
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX (2007) A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39:623–630
Thudi M, Bohra A, Nayak SN, Varghese N, Shah TM, Penmetsa RV, Thirunavukkarasu N, Gudipati S, Gaur PM, Kulwal PL, Upadhyaya HD, KaviKishor PB, Winter P, Kahl G, Town CD, Kilian A, Cook DR, Varshney RK (2011) Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker Loci for chickpea (Cicer arietinum L.). PLoS One 6:e27275
Thudi M, Gaur PM, Krishnamurthy L, Mir RR, Kudapa H, Fikre A, Kimurto P, Tripathi S, Soren KR, Mulwa R, Bharadwaj C, Datta S, Chaturvedi SK, Varshney RK (2014) Genomics-assisted breeding for drought tolerance in chickpea. Funct Plant Biol. 41:1178–1190
Tuberosa R (2012) Phenotyping for drought tolerance of crops in the genomics era. Front Physiol 3:347
Van Ooijen JW, Voorrips RE (2006) JoinMap version4.0: software for the calculation of genetic linkage maps. Wageningen: Plant Research International
Varshney RK, Graner A, Sorrells ME (2005) Genomics-assisted breeding for crop improvement. Trends Plant Sci 10(12):621–630
Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao YL, Srinivasan R, Gaur PM, Siddique KHM, Town CD, Hoisington DA (2009) A comprehensive resource of drought- and salinity-responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.). BMC Genom 10:523
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar’an B, Millan T, Zhang XD, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu CY, Bharti AK, He WM, Winter P, Zhao SC, Hane JK, Carrasquilla-Garcia N, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CLL, Singh NP, Lichtenzveig J, Gali KK, Rubio J, Nadarajan N, Dolezel J, Bansal KC, Xu X, Edwards D, Zhang GY, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR (2013a) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246
Varshney RK, Gaur PM, Chamarthi SK, Krishnamurthy L, Tripathi S, Kashiwagi J, Samineni S, Singh VK, Thudi M, Jaganathan D (2013b) Fast-track introgression of “QTL-hotspot” for root traits and other drought tolerance traits in JG 11, an elite and leading variety of chickpea. Plant Genome 6(3). doi:10.3835/plantgenome2013.07.0022
Varshney RK, Thudi M, Nayak SN, Gaur PM, Kashiwagi J, Krishnamurthy L, Jaganathan D, Koppolu J, Bohra A, Tripathi S, Rathore A, Jukanti AK, Jayalakshmi V, Vemula A, Singh SJ, Yasin M, Sheshshayee MS, Viswanatha KP (2014a) Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor Appl Genet 127:445–462
Varshney RK, Mir RR, Bhatia S, Thudi M, Hu Y, Azam S, Zhang Y, Jaganathan D, You FM, Gao J, Riera-Lizarazu O, Luo M-C (2014b) Integrated physical map with the genetic maps and reference genome sequence for chickpea (Cicer arietinum L.) improvement. Funct Integrat Genomics 14:59–73
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang S, Basten CJ, Zeng ZB (2012) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC
Yu H, Xie W, Wang J, Xing Y, Xu C, Li X, Xiao J, Zhang Q (2011) Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS One 6:e17595
Acknowledgments
Technical assistance from Vinay Kumar is greatly appreciated. The work has been undertaken as a part of Australia–India strategic research fund (AISRF) project funded by Department of Science and Technology (DST) and Tropical Legumes I (TLI) project funded by Bill and Melinda Gates Foundation (BMGF) through CGIAR-Generation Challenge Program (GCP; http://www.generationcp.org/). This work has been carried out as part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of the CGIAR Consortium.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2014_932_MOESM3_ESM.xlsx
The electrophoretic profile of CAPS/dCAPS candidates after PCR and restriction with their respective enzymes (XLSX 139 kb)
438_2014_932_MOESM5_ESM.xlsx
Marker genotyping data from the previous studies included with GBS-SNP markers generated in the present study (XLSX 25 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Jaganathan, D., Thudi, M., Kale, S. et al. Genotyping-by-sequencing based intra-specific genetic map refines a ‘‘QTL-hotspot” region for drought tolerance in chickpea. Mol Genet Genomics 290, 559–571 (2015). https://doi.org/10.1007/s00438-014-0932-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-014-0932-3