
Abstract
There are currently no data available regarding the normal levels of DNA found on the skin of children engaging in routine day to day activities to assist with the forensic interpretation of DNA profiles generated from skin surface swabs. To address this deficit, skin surface swab samples were collected from 12 face/neck sites and 20 body sites on 50 children less than 5 years old. After exclusion of spoilt samples, 60 sets of swabs from 47 children (30 face/neck, 30 body) comprising of 944 individual samples were analysed. The number of alleles observed which could have originated from the child and the number which must have come from another source (non-child) were analysed. The following variables were evaluated: age, kissing, feeding and washing practices, number of contacts and application of cream. Overall, extremely small amounts of non-child DNA were retrieved from skin swabs. Child only (46.3 %) or no DNA at all (18.6 %) was observed for 64.9 % of all swabbed samples. Low levels of non-child DNA (1–5 alleles) were observed on 31.6 % of all swabs tested with only 3.4 % of swabs showing six or more alleles. A great deal of variation between children and between sites in the levels of both child DNA and non-child DNA was observed. A multilevel model, taking account of clustering within children, showed that there was a strong direct association between the amounts of child and non-child DNA observed. There was no relationship between the amount of DNA recovered and the demographic and biographic variables analysed. These background data have the potential to assist the analysis of DNA from the skin of children during criminal investigation.
Similar content being viewed by others


Introduction
DNA technology is increasingly used in forensic investigation, yet there is an absence of data on background DNA levels that may greatly assist the interpretation of DNA profiles. It is widely accepted that biological material containing sufficient DNA for the generation of full or partial short tandem repeat (STR) profiles can be transferred when an individual's skin surface comes into contact with an animate or inanimate object [1–4]. DNA transfer has also been shown to occur during physical assault scenarios in adults, such as manual strangulation [5], with claims that transferred DNA may persist on the touched skin surface for up to 10 days post-contact [6]. Despite the acceptance that detectable levels of DNA can be transferred to an individual's skin surface following physical contact, there is little in the current literature to quantify how much may be present, or if it may be at levels which could confound the interpretation of forensic DNA profiles generated from skin surface samples. Although it has been demonstrated in one small scale study that the skin surface of the adult neck may show ‘background’ levels of non-self DNA due to normal day-to-day activities [7], there is little evidence on how much non-child ‘background’ DNA may already be present on a young child's skin, given that the child may have been handled and kissed repeatedly by their carers and other family members. This study describes DNA levels found on the skin surface of live children aged 0–5 years participating in routine daily activities, and quantifies the background levels of non-child DNA one would expect to find.
Materials and methods
Subjects and sampling
Ethical approval was granted by the multi-centre ethics committee in March 2008 (number 03/9/29), with an approved amendment in July 2008. The study population was a community sample of children aged 0–5 years, living in South Wales, without pre-existing skin disorders or injuries, and in whom there were no child protection concerns. Informed consent was obtained from all participants prior to sample collection.
Sample collection and transportation
Demographic and biographic data were collected, including developmental stage, feeding details (breast or bottle), number of siblings and carers, the number of people who had been in close contact with the child in the 4 h prior to sampling, day-care arrangements and details relating to areas of skin that had been kissed, washed or had cream applied within the preceding 4 h.
Samples were collected from the skin surface of each child from the face/neck at 12 sites and/or body at 20 sites (Fig. 1), using a single moistened cotton swab in a sweeping motion at each site; swabbing was discontinued if the child became distressed. Following sampling, the swab head was removed from the shaft and placed into a 1.5 ml Eppendorf Safe-Lock tube containing 400 μl DNA stabilising solution (DNA Genotek, Ontario, Canada). Reference samples in the form of buccal swabs were collected from the researcher, the child subject and up to four carers or siblings who had contact with the subject in the 4 h preceding sample collection. Samples were stored at room temperature until they were processed.

Areas of the face, neck and/or body that were sampled for each child
Extraction of DNA
DNA was extracted using the QIAamp DNA mini kit (Qiagen, West Sussex, UK) using a method modified for trace-DNA swabs. Whole swab heads were stored submerged in 400 μl DNA stabilisation solution (DNA Genotek, Ontario, Canada), to which 400 μl buffer AL and 20 μl proteinase K was added. The sample tube was mixed and incubated at 56 °C for 10 min. A ‘piggybacking’ stage was then performed, whereby the swab head was transferred to a 0.5 ml sterile plastic tube, with a hole pierced in the bottom. All liquid recovered from the swab head was pooled into the original 1.5 ml tube before continuing with the method as described by the manufacturer. To recover DNA, 60 μl of buffer AE was transferred to the column membrane and was incubated at room temperature for 5 min, then centrifuged at 8,000 rpm for 1 min. The filtrate was then reloaded on to the column, incubated at room temperature for 1 min and centrifuged at 8,000 rpm to collect the DNA. Positive and negative controls were used throughout the process.
Quantification and DNA profiling
DNA quantification was performed using the Quantifiler Duo DNA quantification kit (Applied Biosystems, Foster City, CA, USA) according to manufacturer's instructions. Two no-template controls using 2 μl sterile water in place of template DNA were run in every batch to monitor gross contamination of quantification reagents. Positive and negative DNA extraction controls were also quantified by this method.
Samples containing >0.2 ng/μl DNA were amplified using the SGM Plus polymerase chain reaction (PCR) amplification kit in a total reaction volume of 25 μl using 28 cycles. Whenever possible, 2 ng template DNA was added to each reaction; if less than 2 ng was available, the maximum volume of 10 μl was entered in to the PCR. If, following fragment analysis, partial DNA profiles and/or peaks <35 relative fluorescent units (rfu) were observed, a second run of capillary electrophoresis was performed by loading 2 μl PCR product and using an increased injection time of 10 s instead of the standard 5 s to allow more PCR product to be electrokinetically injected, increasing the sensitivity of the run [8]. For samples containing <0.2 ng/μl, or showing partial or no amplification after 28 PCR cycles or drop-out after the longer injection run, a low copy number (LCN) protocol utilising 34 PCR cycles was carried out [9]. PCR was again performed in a total reaction volume of 25 μl. All samples were amplified at least twice at this stage and a consensus reporting method was used, whereby data were only recorded in the final DNA profile if it appeared in at least two replicate amplifications [9].
DNA profile interpretation
DNA interpretation was based on published guidelines [10] and using thresholds set by internal verification studies. For clarification of our methodology, all peaks above 35 rfu were considered as potential alleles. Alleles observed after standard amplification, using 28 PCR cycles, visualised by standard or longer injection capillary electrophoresis protocols, were always reported in the final DNA profile. If the LCN protocol was used, alleles were only reported in the final DNA profile if they appeared in at least two amplifications, or in a single LCN DNA profile and a previous 28 cycle DNA profile. If the replicate DNA profiles produced after 34 PCR cycles differed by more than six peaks, a third amplification was considered to aid production of a consensus profile which was representative of the DNA present in the sample. Peaks detected in stutter positions were considered as potential alleles if the DNA profile showed evidence of a mixture and the stutter peak height exceeded 20 % of that of the associated allele for standard amplifications and 30 % for LCN amplified DNA. These thresholds were selected following internal validation studies (unpublished data, available on request). For DNA profiles exhibiting characteristics associated with the presence of a minor contributor to a DNA mixture, peaks in stutter positions were considered as potential alleles if they were greater than the average stutter height observed during internal validation experiments for each STR locus, and the height was comparable to other peak heights of the minor DNA profile.
Data analysis
A full reference SGM plus profile was produced for each child volunteer. Alleles present in the sample DNA profile that matched the reference were recorded as ‘child’, and those which could not have come from the child were recorded as ‘non-child’ alleles. An Access database was populated with these data and a bespoke visual basic macro written to organise the data and compare the DNA from each swab with the child DNA and output this in a format appropriate for statistical analysis using Stata ver. 11 and SPSS 18. Because the amounts of DNA, both child and non-child, differed between the swabs subjected to 28 cycles and those which had 34 cycles, results were derived for these two subgroups as well as for the whole set. A multilevel model was fitted to examine the association between the number of non-child alleles observed and the percentage of the child's own DNA profile, modelling sites on the face/neck and body separately [11]. Swabs were nested within children, and the number of non-child alleles was modelled by a Poisson distribution, with robust standard errors to account for possible extra-Poisson variation. A log link function was used, and explanatory variables were the percentage of child DNA profile observed and categorical variables indicating the site of the swab. This model was extended to explore the effects on the number of non-child alleles of child level factors such as age and gender and site-level factors such as having been washed or kissed. In all cases, a P value of 0.05 or less was taken as significant.
Results
Fifty children were recruited for this study. Samples from three children had to be discarded due to sample leakage during transportation. Of the remaining 47, 17 had their face/neck swabbed, 17 had their body swabbed and 13 children had both face/neck and body swabbed at the sites indicated in Fig. 1. This resulted in 30 sets of swabs being collected for the face/neck and 30 sets for the body, a potential of 960 individual samples. Three swabs were not collected due to the intolerance of the child, and 13 were excluded from the final data analysis due to contamination by the sampler during collection, leaving a total of 944 individual samples to be analysed. The allele drop-in rates for the University of Leicester and Northumbria University laboratories were calculated to be 0.04 and 0.01 alleles per locus, respectively. Both of these drop-in rates are below the 0.3 alleles per locus considered as the maximum drop-in rate which can be tolerated for LCN–DNA work to be interpreted with confidence [9]. For DNA extraction controls processed in parallel to buccal swabs, alleles matching one or more of the reference profiles could often be observed in the negative extraction control. For this reason, buccal swabs were processed in separate batches, after all skin-surface swabs had been processed.
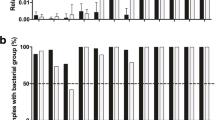
The levels of child and non-child DNA detected varied greatly on a per swab basis. On average, the number of alleles observed in DNA profiles generated from face/neck swabs was greater than for body swabs for both child and non-child DNA. Non-child DNA was consistently low, averaging less than one allele per swab across the entire dataset. Considerable variation in the amounts of both child and non-child DNA was also observed when analysing the data on a per site basis, particularly on the body. Table 1 shows combined results for both 28 and 34 cycle PCR amplification strategies of child and non-child observed on each sampled site. Supplementary data is also provided online to show the breakdown of samples amplified using 28 (Table S1) and 34 cycles (Table S2). Overall, the sites from which most child DNA was recovered were the front and back of the hands, while the fronts of the hands and the cheeks showed the most non-child DNA, although the absolute values were very small. On almost all sites, more child alleles were observed than non-child alleles. More child DNA was found for the 28 cycle data, since the lack of child DNA was one of the triggers for moving to 34 cycles, but the amounts of non-child DNA were comparable for the two datasets.
The data presented in Table 1 suggest that higher levels of non-child DNA were observed on sites, where on average, more than 50 % of the child's own DNA profile was also observed. This was particularly true for data based on 34 cycles. To explore this phenomenon in more detail, the number of non-child alleles observed at each site was compared to the percentage of the child's own DNA profile observed at each site. The data shown in Table 2 suggest that there is an association between the amounts of child and non-child DNA observed. However, it does not take into account the variation in the amount of DNA observed between children or any other demographic or biographic factors that may be influencing the results. Using a multilevel model, for the face/neck and body, the percentage of the child DNA profile observed was associated with the number of non-child DNA alleles that would also be observed (P < 0.0001). This observed association will be validated on a novel dataset, work in progress. The data for 28 and 34 cycle amplification strategies is provided in supplementary Table S3.
The effect of both child and swab level factors was investigated using similar multilevel models. Table 3 shows the numbers of alleles observed and presents the results of the multilevel analysis carried out on the documented variables. The effect size is the ratio of the mean number of non-child alleles observed for the second level for each factor compared to that for the first level. So the ratio for kissing compares the non-child alleles on sites that were kissed to those that were not kissed; the estimate of 1.17 shows that slightly more non-child alleles were found on kissed sites but the confidence interval includes 1, showing that the effect is not significant. In fact, none of the child or site factors shows a significant effect for the whole dataset. The results of fitting this model to the data based on 34 cycles are shown in Table S4 and are broadly similar, not surprising, since this includes the majority of the swabs. The model failed to converge when applied to data based on 28 cycles. This is almost certainly due to the combination of fewer swabs processed using 28 cycles and the smaller numbers of non-child alleles found on these swabs, with some locations having no non-child alleles on any swab. Results on these swabs have, therefore, been omitted.
Discussion
This study demonstrates, for the first time, the background level of DNA retrieved from the skin of active babies and children less than 5 years old. The study highlights that, despite the level of intimate contact between carers and young children, very little non-child DNA is found on their skin. No non-child DNA was observed on 64.9 % of the 944 sites analysed, 1–5 alleles were observed on 31.6 % of all swabs, and more than six non-child alleles were observed for only 3.4 % of all analysed samples. Overall, more DNA was recovered from the face/neck swabs compared to body swabs. The sites with the most child DNA were the front and backs of hands, while the fronts of the hands and the cheeks showed the most non-child DNA. This is perhaps unsurprising due to the level of handling of these areas by carers during routine daily activities.
The finding of low levels of non-child DNA is in keeping with previous work that has been carried out on adult skin surfaces. In 2002, Rutty observed the presence of non-self DNA when investigating the transference of human DNA following manual strangulation. The author commented that these observations may impact upon the interpretation of forensic DNA profile evidence in cases where skin-to-skin contact may have occurred [6]. This work was expanded to include a survey of the background levels of DNA present on the adult neck and showed that non-self DNA were detected on 21 of 120 swabs samples collected from 24 volunteers [7]. In these early articles, it was proposed that non-self DNA may have been present due to secondary or tertiary transfer of DNA that had previously been deposited onto an animate or inanimate intermediate vector. The possibility that some non-self DNA may be present via primary transfer of orally projected DNA should also be considered, as it has been demonstrated that detectable levels of DNA can be projected over 1 m away from an individual when speaking [12].
Aside from these mechanisms of transfer, we also anticipate that DNA may be transferred via skin-to-skin contact during the routine care of babies and young children. It is widely acknowledged that DNA can be transferred from the skin surface to touched objects including the human skin surface [1, 2, 13–15]. More recently, the potential for cell-free nucleic acids to be transferred via skin secretions have also been demonstrated [16]. In addition to the potential for DNA transfer, several authors have commented on the existence of the “shedder-status” of an individual, e.g. [3, 4, 17], yet there does not seem to be any consensus view on how this shedder-status may impact on the interpretation of DNA casework in practice. The lack of unanimity, combined with the overriding opinion that characterisation of shedder-status is influenced by many variables, most notably the passage of time between hand washing and sample deposition [4], and the fact that this research has all been conducted with adult volunteers, led us away from attempting to assign shedder-status in this project.
To ensure that the data generated in this study were truly reflective of background DNA levels present on the child's skin surface, and in accordance with previous studies, external variables that may have influenced the level of self or non-self DNA were not controlled. Volunteers were not requested to perform any activities that would be outside of their daily routine prior to sample collection. Instead, variables which we considered to be potentially influential on the results were recorded by survey. The number of people who had been in close contact with the child in the hours prior to sampling was also recorded. Upon analysis, contact from kissing or breast feeding, the age, gender and developmental level of the child, washing, contacts with multiple people or skin cream application did not appear to significantly alter the amount of non-child DNA we observed.
There was a significant association between the amount of child and non-child DNA recovered; when non-child DNA was observed, it tended to be on swabs where a greater proportion of the child's own DNA profile was also observed. As none of the recorded variables showed a significant effect on the number of non-child alleles detected, they cannot explain this relationship. Initially, we attempted to identify the origin of non-child alleles observed in the DNA profile by comparison to reference DNA profiles from individuals who had been in physical contact with the child in the 4-h period prior to sample collection. This approach was confounded by the unavailability of such individuals to provide a buccal swab at the time of sample collection. A decision was taken to record alleles observed in the DNA profile as ‘child’ or ‘non-child’, without attempting to identify the source of the non-child alleles. This did not meaningfully affect our data analysis, as the intention was to determine the ‘background’ levels of DNA present on the skin surface of young children. We confirmed that background non-child DNA levels are very low. This supports the potential for future exploration of the validity of DNA sampling in children.
As young children were recruited to this study, it was neither appropriate nor practical to use full PPE, i.e. scene suit, face-mask, mobcap, overshoes and gloves. We have previously confirmed that little sampler contamination occurred despite this omission [18]. In this study, 13 of the 960 collected samples had been contaminated by the sample collector and were excluded from the analysis. Initially, some contamination of sample collection negative controls was observed. Upon analysis, it was discovered that alleles in common with the sample collector could often be observed in these negative controls, but not in the DNA profiles for the samples collected from the skin surface. Following discussion, we discovered that the sample collector was handling the control swabs close to her body, whilst handling the actual samples at arm's length. When all samples, including the negative control swabs, were handled at arm's length, this problem was resolved. If this methodology was to be adapted for clinical use, on live children, we would advocate the minimal PPE approach but would advise that appropriate training was undertaken to minimise the risk of further contamination.
Conclusions
We have reported for the first time levels of ‘background’ DNA found on the skin of infants and very young children, highlighting that, despite intimate contact with carers and siblings, levels of non-child DNA found are extremely low. These results have the potential to provide a baseline to any forensic assessment of DNA samples taken from a child, where injuries are suspected to have arisen from a physical blow or bite from a third party. We have shown an interesting, albeit unexplained, association between the amount of child and non-child DNA retrieved, which merits further exploration. For this approach to be fully utilised, it would be necessary to further our understanding of the mechanisms of DNA transfer involving skin cells, body fluids and cell-free nucleic acids during both social and anti-social acts, to both inanimate and animate surfaces, under varying test conditions [14, 15, 19]. This important baseline data provides a first step toward the potential use of DNA techniques in evaluating suspected child abuse in children who are too young to provide a history of any injuries they have sustained.
References
Daly DJ, Murphy C, McDermott SD (2012) The transfer of touch DNA from hands to glass, fabric and wood. Forensic Sci Int Genet 6(1):FS1–FS46. doi:10.1016/j.fsigen.2010.12.016
van Oorschot RA, Jones MK (1997) DNA fingerprints from fingerprints. Nature 387(6635):767
Lowe A, Murray C, Whitaker J, Tully G, Gill P (2002) The propensity of individuals to deposit DNA and secondary transfer of low level DNA from individuals to inert surfaces. Forensic Sci Int 129(1):25–34
Phipps M, Petricevic S (2007) The tendency of individuals to transfer DNA to handled items. Forensic Sci Int 168(2-3):162–168. doi:10.1016/j.forsciint.2006.07.010
Wiegand P, Kleiber M (1997) DNA typing of epithelial cells after strangulation. Int J Legal Med 110(4):181–183
Rutty GN (2002) An investigation into the transference and survivability of human DNA following simulated manual strangulation with consideration of the problem of third party contamination. Int J Legal Med 116(3):170–173
Graham EAM, Rutty GN (2008) Investigation into "normal" background DNA on adult necks: implications for DNA profiling of manual strangulation victims. J Forensic Sci 53(5):1074–1082. doi:10.1111/j.1556-4029.2008.00800.x
Forster L, Thomson J, Kutranov S (2008) Direct comparison of post-28-cycle PCR purification and modified capillary electrophoresis methods with the 34 cycle "low copy number" (LCN) method for analysis of trace forensic DNA samples. Forensic Sci Int Genet 2(4):318–328. doi:10.1016/j.fsigen.2008.04.005
Gill P, Whitaker J, Flaxman C, Brown N, Buckleton J (2000) An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci Int 112(1):17–40
Whitaker JP, Cotton EA, Gill P (2001) A comparison of the characteristics of profiles produced with the AMPFlSTR SGM Plus multiplex system for both standard and low copy number (LCN) STR DNA analysis. Forensic Sci Int 123(2–3):215–223
Goldstein H (2003) Multilevel Statistical Models. 3rd Edition edn. Edward Arnold, London
Port N, Bowyer VL, Graham EAM, Batuwangala M, Rutty GN (2005) How long does it take a static speaking individual to contaminate the immediate environment? Forensic Sci Med Pathol 2(3):157–163
Alessandrini F, Cecati M, Pesaresi M, Turchi C, Carle F, Tagliabracci A (2003) Fingerprints as evidence for a genetic profile: morphological study on fingerprints and analysis of exogenous and individual factors affecting DNA typing. J Forensic Sci 48(3):586–592
Goray M, Eken E, Mitchell RJ, van Oorschot RAH (2010) Secondary DNA transfer of biological substances under varying test conditions. Forensic Sci Int Genet 4(2):62–67. doi:10.1016/j.fsigen.2009.05.001
Goray M, Mitchell RJ, van Oorschot RAH (2010) Investigation of secondary DNA transfer of skin cells under controlled test conditions. Legal Med 12(3):117–120. doi:10.1016/j.legalmed.2010.01.003
Quinones I, Daniel B (2005) Cell free DNA as a component of forensic evidence recovered from touched surfaces. Forensic Sci Int Genet. doi:10.1016/j.fsigen.2011.01.004
Balogh MK, Burger J, Bender K, Schneider PM, Alt KW (2003) STR genotyping and mtDNA sequencing of latent fingerprint on paper. Forensic Sci Int 137(2–3):188–195
Maguire S, Ellaway B, Bowyer VL, Graham EA, Rutty GN (2008) Retrieval of DNA from the faces of children aged 0–5 years: a technical note. J Forensic Nurs 4(1):40–44. doi:10.1111/j.1939-3938.2008.00005.x
Goray M, van Oorschot RAH, Mitchell JR (2012) DNA transfer within forensic exhibit packaging: potential for DNA loss and relocation. Forensic Sci Int Genet 6(2):158–166. doi:10.1016/j.fsigen.2011.03.013
Acknowledgements
We wish to thank the Medical Research Council for supporting this project.
Conflict of interests
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Graham, E.A.M., Watkins, W.J., Dunstan, F. et al. Defining background DNA levels found on the skin of children aged 0–5 years. Int J Legal Med 128, 251–258 (2014). https://doi.org/10.1007/s00414-013-0906-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00414-013-0906-8