
Abstract
The heart’s rhythm is initiated and regulated by a group of specialized cells in the sinoatrial node (SAN), the primary pacemaker of the heart. Abnormalities in the development of the SAN can result in irregular heart rates (arrhythmias). Although several of the critical genes important for SAN formation have been identified, our understanding of the transcriptional network controlling SAN development remains at a relatively early stage. The homeodomain transcription factor Shox2 is involved in the specification and patterning of the SAN. While the Shox2 knockout in mice results in embryonic lethality due to severe cardiac defects including improper SAN development, Shox2 knockdown in zebrafish causes a reduced heart rate (bradycardia). In order to gain deeper insight into molecular pathways involving Shox2, we compared gene expression levels in right atria of wildtype and Shox2 −/− hearts using microarray experiments and identified the LIM homeodomain transcription factor Islet1 (Isl1) as one of its putative target genes. The downregulation of Isl1 expression in Shox2 −/− hearts was confirmed and the affected region narrowed down to the SAN by whole-mount in situ hybridization. Using luciferase reporter assays and EMSA studies, we identified two specific SHOX2 binding sites within intron 2 of the ISL1 locus. We also provide functional evidence for Isl1 as a transcriptional target of Shox2 by rescuing the Shox2-mediated bradycardia phenotype with Isl1 using zebrafish as a model system. Our findings demonstrate a novel epistatic relationship between Shox2 and Isl1 in the heart with important developmental consequences for SAN formation and heart beat.
Similar content being viewed by others


Introduction
During embryogenesis most cells have to become specialized into one of many various cell types that build a whole organism. These patterning and differentiation processes are ensured by the temporal and spatial activation of different transcription factors [50]. Homeobox genes, for example, regulate numerous processes during embryogenesis including heart development. Complex transcriptional regulation networks are important in many aspects of heart cell lineage development and morphogenesis [46].
The cardiac conduction system consists of electrically coupled cardiomyocytes that are responsible for impulse propagation and essential for the rhythmic and coordinated contraction of the heart. A specialized group of cells in the sinoatrial node (SAN) located at the junction of the superior caval vein and right atrium generates the electric impulse to activate the atrial myocardium (also termed primary pacemaker). Together with specialized cells in the atrioventricular node (termed secondary pacemaker) and conduction fibers in the interventricular septum, they form the cardiac conduction system [7, 19]. Abnormalities in the cardiac conduction pathway are responsible for various forms of arrhythmias [41, 44, 58]. Sinus node dysfunction, for example, is associated with sinus bradycardia, atrial tachyarrhythmias, sinus pause or arrest and sinoatrial exit block [10]. Although the morphology, cellular components and electrophysiological properties of the SAN are rather well described [34, 38], knowledge of pathways and transcriptional regulation of the SAN formation are poorly understood. Unraveling the pathways that control pacemaker development and function is of crucial relevance to our understanding of pathologies associated with cardiac conduction.
So far, several transcription factors of the homeobox- and T-box gene families have been shown to be involved in cardiac conduction system specification, patterning, maturation, and function [17, 37]. Among these genes is the paired-related homeodomain transcription factor Shox2, which is known to play a crucial role in early cardiac formation, particularly in SAN development and specification [2, 11, 12, 30, 42]. Shox2 expression can first be detected in the posterior region of the primitive heart tube at murine embryonic stage E8.5 and as development progresses, Shox2 is specifically expressed in the sinus venosus myocardium comprising the SAN and the venous valves [2, 12]. These Shox2 expressing domains develop from myocardium that is added to the venous pole of the developing heart and contributes to the second heart field lineage [5, 52]. Newly recruited myocardium that is added to the arterial pole is divided into the anterior or secondary heart field, while the myocardium added to the venous pole is referred to as the posterior heart field [15]. Loss of function studies revealed an essential role for Shox2 in the normal anlage of the posterior heart field myocardium [2, 12]. Shox2 −/− mice die between E11.5 and E17.5 due to heart failure and vascular defects. In particular, the sinus venosus myocardium including the SAN and venous valves show hypoplasia. Moreover, Shox2 has a crucial role in pacemaker function indicated by severe bradycardia with intermittent sinus exit block after morpholino-mediated knockdown in zebrafish embryos and markedly reduced heart beat rates of isolated Shox2 −/− hearts [2, 12]. To investigate the molecular processes underlying Shox2 function, different marker gene expression analyses have been performed. The aberrant expression of Cx40, Cx43, Nppa, and Nkx2.5 within the SAN of Shox2 mutant hearts as well as the decreased expression of the SAN-specific marker genes Tbx3 and Hcn4 has indicated an abnormal differentiation of pacemaker cells [2, 12]. It has also been shown that Shox2 prevents the SAN from atrialization by repressing Nkx2.5 expression [11, 12]. Recently, a further link between Tbx5, Shox2 and Bmp4 could be established in the developing heart, demonstrating that Tbx5 signaling in the cardiac pacemaker region controls the expression of Shox2, which in turn regulates Bmp4 in a direct manner [42]. It has also been reported that Pitx2c represents an important susceptibility gene for atrial arrhythmias by suppressing left-sided sinus node formation via direct repression of Shox2 [25, 53]. Taken together, these findings illustrate the importance of Shox2 function in SAN development and highlight the need for further elucidation of the molecular networks involved.
The aim of our current study was to identify Shox2 target genes during heart development. We used expression analysis to compare the transcriptomes in right atria of wildtype and Shox2 −/− mouse hearts, and Islet1 (Isl1) was identified as a direct transcriptional target. We have examined putative regulatory elements in the human ISL1 gene and show a pivotal role for sequences within intron 2 in activating transcription. Furthermore, a functional link between Shox2 and Isl1 in vivo was demonstrated using zebrafish as a model by rescuing the Shox2-mediated bradycardia via ectopic expression of Isl1.
Methods
Animals and tissue samples
Shox2 −/− mice were generated and genotyped as previously described [2]. For maximal reproduction, we crossed our mice into the CD-1 outbred strain. To improve specificity of the microarray analysis, we dissected the right atria of E11.5 hearts that comprise the Shox2 expressing sinus venosus myocardium and venous valves. For total RNA isolation by TRIzol® (Invitrogen), samples from embryonic right atria (E11.5) of the same genotype (wildtype and Shox2 −/−) were pooled. For whole-mount in situ hybridization, complete hearts were dissected from E11.5 and E12.5 embryos and fixed overnight in 4 % paraformaldehyde at 4 °C.
Microarray analysis
Gene expression profiling was performed using the GeneChip Mouse Gene 1.0 ST array from Affymetrix (Santa Clara, CA) according to the manufacturer’s protocol. For each genotype (wildtype and Shox2 −/−), RNA from 6 right atria from embryos of 2 independent pregnancies was pooled. Purity and quality of isolated RNA were assessed by the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and showed a RIN >9. 200 ng RNA were used for production of end-labeled biotinylated ssDNA and subsequently hybridized to the arrays. Statistical comparisons of chip data based on ANOVA were performed using the software package JMP Genomics, version 4.0 from SAS (SAS Institute, Cary, NC, USA). Briefly, values of perfect-matches were log transformed, quantile normalized and fitted with log-linear models, with probe ID and genotype considered to be constant. Custom CDF version 14 with Entrenz gene-based gene definitions (http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/genomic_curated_CDF.asp) different from the original Affymetrix probe set definitions was used to annotate the arrays. The microarray data were deposited in the NCBI GEO database with accession number GSE39924.
Quantitative RT-PCR
cDNA was synthesized using the SuperScript™ First-Strand Synthesis System for RT-PCR (Invitrogen). Quantitative RT-PCR (qRT-PCR) was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems) using SYBR Green ROX dye (Thermo Scientific). Primer sets used are presented in Supplemental Material (Table S1).
Generation of plasmid constructs
Cloning of the murine Isl1 in situ probe, the zebrafish shox2 and isl1 expression constructs, the human SHOX2a expression construct and generation of the ISL1 luciferase reporter constructs are described online in the Supplemental Material.
In situ hybridization
Shox2 −/− mice have been described previously [2]. Whole-mount in situ hybridization on embryonic mouse hearts was performed as reported [16]. Specific digoxygenin- or fluorescein-labeled RNA probes were as follows: Isl1 (see Supplemental Material) and Hcn4 (kindly provided by B. Bruneau, Gladstone Institute of Cardiovascular Disease, USA).
Immunohistochemistry
E11.5 embryos were fixed in 4 % paraformaldehyde at 4 °C overnight, embedded in OCT and transversely cryosectioned (10 μm). Tbx18 and Hcn4 stainings were enhanced using the ABC (Avidin–Biotin-Complex) reagent from Vector Laboratories (VECTASTAIN Elite Peroxidase ABC-Kit) and the TSA (Tyramide Signal Amplification) system (NEL741, Perkin Elmer) according to the manufacturers’ instructions. For double staining (Isl1/Tbx18 or Isl1/Hcn4), the respective fluorescent secondary antibody was added either during the biotinylated antibody incubation or in a second step afterwards. The following primary antibodies were used: mouse monoclonal antibody against Isl1 (1:100; 39.4D5, Developmental Studies Hybridoma Bank), goat polyclonal antibody against Tbx18 (1:250; C-20, Santa Cruz) and rabbit polyclonal antibody against Hcn4 (1:500; APC-052, Alomone). Alexa Fluor 568 rabbit anti-mouse (1:800; A-11061, Invitrogen), biotinylated horse anti-goat (1:500; BA-9500, Vector Laboratories) and biotinylated goat anti-rabbit (1:500; 550338, BD Pharmingen) were used as secondary antibodies. Nuclei were counterstained with Hoechst (Invitrogen). Imaging was carried out on a Nikon 90i upright epi-fluorescence microscope with a Nikon DS-Qi1 mc camera.
Cell culture, transfection and luciferase assay
HEK 293 cells were cultured at 37 °C in DMEM medium containing high glucose, supplemented with 10 % fetal calf serum and antibiotics. For luciferase assay analysis, the cells were cotransfected in triplicate with different constructs using polyethylenimine (Sigma-Aldrich). 24 h after transfection, luciferase activity was determined and normalized to Renilla luciferase activity with a dual luciferase assay kit (Promega). Experiments were repeated at least three times in triplicate with consistent results and representative data are shown.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed using standard protocols. For the binding reaction, 32P-labeled, double stranded ISL1 oligonucleotides (Oligo1 (+2364/+2418), Oligo2 (+2404/+2458), Oligo3 (+2447/+2502), Oligo4 (+2496/2561); Supplemental Material Table S1), were used together with purified, bacterially expressed recombinant GST-SHOX2 protein [3].
Zebrafish embryos and microinjections
Care and breeding of zebrafish, Danio rerio, were as described previously [55]. For all plasmid and morpholino injection procedures, the TE4/6 wildtype strain was used.
Morpholino-modified antisense oligonucleotides (MO; Gene Tools) were directed against the translational start site (shox2-MO [5′-ACGCTGTAAGTTCTTCCATCACTGC-3′]) of zebrafish shox2, the splice donor site of exon 2 (isl1-MO [5′-TTAATCTGCGTTACCTGATGTAGTC-3′]) of zebrafish isl1 and the translational start site (isl1-MO2 [5′-GATCCCCCATGTCTCCCATGTCAAG-3′] of zebrafish isl1. A shox2-MO directed against the splice donor site of intron 3 causing the identical bradycardia phenotype has been described previously [2]. shox2 and isl1 antisense oligonucleotides or a standard control oligonucleotide (MO-control), diluted in 0.2 mol/liter KCl, were microinjected into one-cell-stage zebrafish embryos [20]. For rescue experiments 0.32 ng of pDestTol2CG2-shox2 or pDestTol2CG2-isl1 was microinjected into one-cell-stage embryos directly after injection of 2.8 ng of shox2-MO.
For histology, embryos were fixed in 4 % paraformaldehyde and embedded in JB-4 (Polysciences, Inc). Then, 5-μm sections were cut, dried, and stained with hematoxylin and eosin [43].
For immunoblotting, monoclonal mouse IgG1 anti-rat islet1 antibody (Developmental Studies Hybridoma Bank, DSHB, University of Iowa) and polyclonal anti pan-cadherin (Abcam) were used. Proteins were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membrane. Blots were probed with the mentioned antibodies, signals were detected by chemiluminescence (monoclonal anti-rabbit-Hrp and anti-mouse-Hrp) and imaging was performed with Image Quant LAS4000mini (GE Healthcare). Quantification of the immunoblot was performed using the Image Quant TL software.
Whole-mount antisense RNA in situ hybridization was carried out as described [21] using a digoxigenin-labeled antisense probe for zebrafish shox2 and islet1. For immunostaining, zebrafish embryos were fixed in Dent’s fixative and a monoclonal anti-acetylated tubulin antibody (Sigma) was used [40].
Statistical analysis
Data are presented as mean ± SEM from at least three independent experiments. Comparisons between experimental groups were performed using paired t test or 1-way ANOVA. Differences were considered significant if they showed a value of P < 0.05.
Results
Shox2 has regulatory effects on Isl1 expression in the developing sinoatrial node
To identify novel transcriptional targets of Shox2, we performed microarray analysis and compared gene expression levels in right atria of wildtype and Shox2 −/− hearts. Only right atria comprising the Shox2 expressing sinus venosus myocardium and venous valves were dissected from murine E11.5 hearts to increase the specificity of the array analysis. Genes were considered to be up- or downregulated if they showed log2 ratios of ≥0.35 or ≤−0.4 with a significance of P ≤ 0.0001. Using these selection criteria, we identified 321 genes with significantly altered gene expression (80 up- and 241 downregulated genes). As expected, Shox2 was among the highest negatively regulated genes. Classification of the 321 genes into gene families using the Gene Set Enrichment Analysis (GSEA) revealed that the highest proportion of regulated genes belongs to the group of transcription factors. Among these, the LIM homeodomain transcription factor Isl1 showed a reduced expression in right atria of Shox2 −/− embryos.
To verify Isl1 as a putative target gene, we carried out qRT-PCR analyses using right atrial tissue samples of wildtype and Shox2 knockout embryos and confirmed the downregulation of Isl1 expression in a second independent pool of embryonic heart tissue (Fig. 1A). To further investigate Isl1 as a putative target gene of Shox2, we compared the spatial expression of both genes and found that Shox2 and Isl1 expression overlap in embryonic mouse hearts. Isl1 expression was detected in the SAN of wildtype hearts, which corresponds to the area where Shox2 normally is expressed (Fig. 1B). Whole-mount in situ hybridization on isolated E11.5 wildtype and knockout hearts demonstrated that Isl1 transcripts are completely absent in the SAN region of Shox2 mutants (Fig. 1C b’), while normal expression was observed in the outflow tract (Fig. 1C b). The lack of Isl1 expression in the SAN, but unaffected Isl1 expression in the outflow tract of Shox2-null mice could be also confirmed at embryonic stage E12.5 (Fig. S1A). In situ analyses revealed a complete loss of Isl1 expression in the right atrium (SAN) of knockout hearts, suggesting that the residual Isl1 expression detected in Shox2 knockout tissue by qRT-PCR and microarray experiments is likely due to some remaining outflow tract tissue following microdissection. Quantitative expression analysis using outflow tract tissue samples showed high and consistent Isl1 expression levels in both wildtype and Shox2 knockout embryos (Fig. S1B).

Shox2 deficiency impairs Isl1 expression. A qRT-PCR analysis using right atrial tissue of E11.5 wildtype and Shox2 −/− mouse hearts. Shox2 knockout results in 34 % reduction of Isl1 mRNA levels in the right atrium of the heart. All results were normalized to Sdha (succinate dehydrogenase complex, subunit A) and Hprt1 (hypoxanthine phosphoribosyltransferase 1) mRNA values. (** P = 0.0058; n = 21 right atria from 6 different pregnancies and 6 independent experiments). B Coexpression of Shox2 and Isl1 in the embryonic mouse heart. Sections after whole-mount in situ hybridization on E11.5 wildtype mouse hearts using Shox2 (upper section) and Isl1 (lower section) RNA probes. Murine Shox2 and Isl1 are coexpressed specifically in the SAN region of the developing heart (indicated by black arrowheads). C Isl1 expression is absent in the SAN of Shox2 −/− embryos. Whole-mount in situ hybridization on E11.5 wildtype (a) and Shox2 −/− (b) mouse hearts using an Isl1 RNA probe. Both, ventral (a, b) and dorsal (a’, b’) views are shown. Murine Isl1 is expressed in the OFT (a) and in the SAN (a’) of the developing heart in wildtype embryos. In the Shox2 −/− mouse heart, Isl1 expression is still present in the OFT (b) but completely absent in the SAN (b’), where Shox2 and Isl1 expression domains overlap in the wildtype heart. RA right atrium, LA left atrium, RV right ventricle, LV left ventricle, OFT outflow tract, SAN sinoatrial node
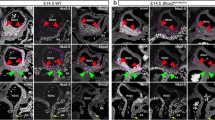
To validate that the loss of Isl1 expression in the SAN of Shox2 knockout hearts is not due to incorrect formation of SAN tissue, we examined the expression of SAN markers in wildtype and knockout hearts using in situ hybridization and immunohistochemistry. While Tbx18 is mainly expressed in the SAN head, Hcn4 is a very prominent marker of the complete developing SAN [45, 56]. Double in situ hybridization experiments using an Isl1 antisense probe together with Hcn4 showed coexpression in the SAN of E12.5 and E13.5 wildtype hearts (Fig. 2A a, a’, c, c’). In Shox2 −/− mouse hearts Isl1 expression is completely absent in this region, while Hcn4 transcripts are still detectable (Fig. 2A b, b’, d, d’). Double immunostainings revealed that Hcn4 and Tbx18 colocalize with Isl1 in the SAN of E11.5 wildtype embryos (Fig. 2B a, g). While both markers could be detected in the SAN of wildtype and knockout hearts (Fig. 2B c, f, i, l), Isl1 staining is not detectable in this region in Shox2 −/− embryos (Fig. 2B e, k). Together, these stainings confirm the loss of Isl1 expression in the SAN of Shox2 mutants on transcript and protein level at different embryonic stages, while the presence of Hcn4 and Tbx18 confirm that the SAN is formed. The loss of Isl1 expression is thereby very likely linked to Shox2 deficiency and does not constitute an indirect effect due to disrupted development of SAN tissue in knockout embryos.

Double staining of wildtype and Shox2 −/− embryos. A Double whole-mount in situ hybridization with corresponding sections (50 μm) on E12.5 (a, b) and E13.5 (c, d) wildtype and Shox2 −/− mouse hearts using Isl1 and Hcn4 RNA probes. Murine Isl1 (blue staining) and Hcn4 (red staining) are expressed in the SAN (a, a’, c, c’) of the developing heart in wildtype embryos (indicated by black arrowhead). In the Shox2 −/− mouse heart Isl1 expression is completely absent in the SAN (b, b’, d, d’), which represents the overlapping expression domains of Shox2 and Isl1 in the wildtype heart. Note that Hcn4, another marker of the SAN region, is still expressed in Shox2 −/− hearts, even if the expression is somewhat reduced (b, b’, d, d’). B Transverse sections (10 μm) of E11.5 wildtype (a–c; g–i) and Shox2 −/− (d–f; j–l) embryos were stained with Isl1 (red) and the SAN marker Hcn4 or Tbx18 (green). Nuclei are counterstained with Hoechst (blue). Isl1 and Hcn4 expression clearly overlaps in the SAN region of wildtype embryos (a; white arrowhead). Hcn4 is still detectable in the SAN of Shox2 −/− embryos (f), whereas no Isl1 staining is visible in this region (e). Isl1 and Tbx18 expression clearly overlaps in the SAN head of wildtype embryos (g; white arrowhead). Tbx18 is still detectable in the SAN of Shox2 −/− embryos (l), whereas no Isl1 staining is visible in this region (k). RA right atrium, LA left atrium, RV right ventricle, LV left ventricle, SAN sinoatrial node. Scale bars: a–f 1,000 μm; g–l 500 μm
SHOX2 binds to regulatory elements in an intronic region of the ISL1 gene
To further study the transcriptional control mechanism of ISL1 by SHOX2, we investigated specific binding sites within the ISL1 locus. As a first step, we performed multispecies conservation analysis for the human ISL1 locus to identify evolutionary conserved regions. Conservation of DNA sequences provides a route for identifying functional elements in genomes [32]. Sequence comparison of human, frog, chicken, and mouse Isl1 revealed several highly conserved regions (Fig. S2A). Using luciferase assays, we functionally characterized these highly conserved elements. The reporter constructs used are depicted as black boxes below the conservation plot in Supplemental Material Fig. S2A and are referred to as ISL1-reporter-1 to ISL1-reporter-5. ISL1-reporter-3 and -4 include regulatory elements previously identified downstream of the translational stop codon [22, 23], while ISL1-reporter-1 contains a recently published minimal promoter of the Isl1 gene [31]. Cotransfections of the different ISL1 reporter constructs along with a SHOX2a expression plasmid revealed a strong increase in luciferase activity only for ISL1-reporter-2, compared to the basal level of the respective reporter alone (Fig. S2B). By transfection of serial gene deletion constructs (genomic locations of the constructs are depicted in Fig. 3A), we confined the SHOX2 transcriptional regulatory element to a 184-bp region located in the second intron of ISL1, corresponding to ISL1-reporter-2del 3 (Fig. 3A, B). To identify the precise SHOX2 binding site, EMSA was performed using four different oligonucleotide probes (Oligo1–Oligo4; Fig. S3A) that cover the respective intronic ISL1 sequence. We obtained a shift with both Oligo1 (strong binding) and 3 (weaker binding), but not with Oligo2 and 4 (Fig. 3C, Fig. S3B). Competitive EMSAs demonstrated that only high excess (150-fold) of unlabeled Oligo3 (SpC3) competitively affects binding of Oligo1 to SHOX2 (Fig. S4A, left panel), while already a low molar excess (50-fold) of unlabeled Oligo1 (SpC1) competitively inhibits binding of labeled Oligo3 to SHOX2 (Fig. S4A, right panel). Our luciferase assays also show that the sequence encompassing Oligo1 alone is not sufficient to activate ISL1 by SHOX2 (as this region was also partly included in the ISL1-reporter-2del 2 construct which did not show an increase in luciferase activity upon cotransfection with SHOX2) (Fig. 3B), suggesting that both sequences (encompassing Oligo1 and Oligo3) are necessary to activate the ISL1 gene by SHOX2. Oligo1 and Oligo3 both include AATT-palindromic sequences, which were mutated to CCGG (Oligo1 mut and Oligo3 mut) to confirm sequence specific binding. No shift was obtained using the mutated Oligos, confirming that the AATT motif is essential for SHOX2 binding (Fig. 3C). In addition, competition assays revealed that an excess of unlabeled wildtype Oligos (SpC1 and SpC3) reduce the DNA binding ability of SHOX2, whereas excess of mutant Oligos (MuC1 and MuC3) do not affect the binding (Fig. S4B).

SHOX2 binds to regulatory elements within the ISL1 gene. A Schematic drawing showing the position of the luciferase reporter construct sequences within the genomic region of ISL1 and the probes generated for EMSA. Boxes and lines represent exons and introns, respectively. EMSA probes to which SHOX2 binds are indicated in red. B SHOX2 increases the transcriptional activity of the ISL1-reporter-2, as evaluated by luciferase assay. The responsive reporter element was further narrowed down using deletion (del) constructs. HEK 293 cells were transiently cotransfected with the indicated ISL1 reporter constructs or the empty pGL3 basic vector together with a SHOX2 expression plasmid or an empty vector (pST-Blue1) and 24 h after transfection, luciferase activity was determined. Experiments were repeated at least three times in triplicate with consistent results and representative data are shown. C EMSA revealed specific binding using Oligo1 and Oligo3 together with a GST-SHOX2 fusion protein (shifted band is marked by an arrowhead). Precise binding sites were identified by the use of mutated probes (Oligo1 mut and Oligo3 mut), with 4 nucleotides exchanged (AATT ↔ CCGG). Free oligo is indicated by an arrow. Representative data from three independent experiments with consistent results are shown
Bradycardia caused by shox2 deficiency depends on isl1 expression
To functionally assess whether Shox2 and Isl1 act in the same genetic pathway, we injected zebrafish embryos with morpholinos (MO). When injecting either 2.8 ng of shox2-MO or isl1-MO, 82.0 ± 2.6 % (n = 175; 2 independent experiments) and 79.1 ± 0.3 % (n = 182; 2 independent experiments) of injected embryos, respectively, displayed pericardial edema (Fig. 4A asterisk) and a severely reduced heart rate (Fig. 4B). Embryonic heart morphogenesis proceeds normally in shox2-MO and isl1-MO injected zebrafish. By 72-h post fertilization (hpf), morphant hearts consist of two heart chambers, atrium and ventricle, with properly developed myocardial and endocardial cell layers. The similarity of phenotypes between shox2 and isl1 deficiency in the fish raised the possibility that a close functional interaction between these two genes exists. To address whether the observed cardiac dysfunction caused by shox2 deficiency could depend on isl1 function, we examined the ability of isl1 to compensate the morpholino-mediated loss of shox2 function by transient rescue experiments. DNA constructs expressing isl1 and shox2 under the control of the cardiac myosin light chain 2 (cmlc2) promoter [18, 33], which drives myocardial-specific expression were used for the rescue experiment. The cmlc2 promoter also drove expression of GFP, which allowed monitoring of cardiomyocyte-specific expression (Fig. S5A). We coinjected 0.32 ng of the isl1-expression construct along with 2.8 ng shox2-MO, which was sufficient to induce the bradycardia phenotype and subsequently measured the heart rate of GFP positive embryos after 72 hpf. Myocardial-specific expression of isl1 could significantly rescue the reduced heart rate in shox2 morphant embryos. As a control, injection of the isl1- or shox2-expression construct alone did not cause any changes in embryonic heart rate (Fig. 4C). Histological sections of shox2-MO injected hearts rescued by isl1 overexpression show normal development of the heart without formation of pericardial edema (Fig. 4D), compared to those of shox2-MO injected embryos alone (Fig. 4A). In addition, we could show a downregulation of isl1 protein levels in embryos injected with shox2-MO (Fig. S5B). Both proteins, shox2 and isl1, are highly expressed during neural development and isl1 plays an important role during differentiation of neurons in the brain. To exclude that shox2 deficiency also affects differentiation of isl1 positive neurons, we analyzed the isl1 expression after shox2 knockdown by whole-mount in situ hybridization. Isl1 expression in the brains of shox2 morphants is indistinguishable from wildtype embryos (Fig. S6A and B). On the other hand, isl1 deficient embryos show a normal expression of shox2 in the head region (Fig. S6C and D). Furthermore, we performed immunostaining using acetylated Tubulin as neuronal marker (Fig. S6E and F). While the heart rate was reduced after shox2-MO injection, no alteration could be detected in neural development. Together, these results indicate an epistatic relationship and a direct functional link between shox2 and isl1 in the pacemaking system of zebrafish embryos.

shox2-MO-mediated bradycardia can be rescued by isl1 overexpression. A Injection of 2.8 ng shox2 and isl1 antisense morpholinos (shox2-MO and isl1-MO) leads to pericardial edema and pericardial blood congestion due to reduced heart rates in zebrafish embryos. Lateral views of WT, shox2-MO and isl1-MO injected embryos 72 h post fertilization (hpf). Hematoxylin and eosin staining of sagittal histological sections of WT (left), shox2-MO (middle) and isl1-MO (right) morphant hearts 72 hpf. Endocardial and myocardial layers of ventricle and atrium are defined, and heart chambers are separated by an atrioventricular ring. B Heart rates of shox2 and isl1 morphants are decreased by 72 hpf. Injection of shox2-MO leads to sinus bradycardia with heart frequencies of 130 beats per min (bpm) compared to WT embryos with 172 bpm. isl1 deficient embryos exhibit severe sinus bradycardia and frequent pauses in the cardiac contraction with heart frequencies of 59 bmp. (***P < 0.001). C shox2-MO-mediated bradycardia is partially rescued by cardiomyocyte-specific expression of isl1. The mean heart rate of WT embryos 72 hpf amounts to 160 bpm. After shox2 knockdown (2.8 ng shox2-MO) the heart rate is reduced to 114 bpm. Cardiomyocyte-specific overexpression of shox2 or isl1 alone (0.32 ng) does not affect the heart beat. Coinjection of 0.32 ng shox2 or isl1 plasmid DNA along with 2.8 ng shox2-MO results in significantly increased heart frequencies (134 and 137 bmp) compared to shox2-MO-injected embryos (114 bpm) at 72 hpf (****P < 0.001; n = 32–40 per condition). D Hematoxylin and eosin staining of sagittal histological heart sections of WT (left) and shox2-MO embryos rescued by isl1 overexpression (right) 72 hpf revealed that the rescued hearts, compared to those of shox2-MO injected embryos (A, middle picture), show a phenotype very similar to the WT (left). WT wildtype, asterisk pericardial edema, double asterisks no pericardial edema
Discussion
During embryogenesis, the proper development of the cardiac pacemaker is essential for a coordinated heart beat. Defects in the transcriptional network controlling SAN development may cause dysfunction of the primary pacemaker resulting in severe arrhythmias. The elucidation of the molecular mechanisms and key regulators involved in these developmental processes is therefore crucial for our understanding of arrhythmia-related diseases.
The LIM homeodomain transcription factor Isl1 is a prominent marker of second heart field derivatives comprising the right ventricle, outflow tract, inflow tract with the SAN and parts of the atria and plays a crucial role in cardiogenesis [6, 49, 51]. SAN development is initiated in the presence of Isl1 expressing second heart field-derived cells [35, 48, 51]. Homozygous loss of Isl1 results in embryonic lethality between E10.5 and E11.0 due to severe cardiac abnormalities. Besides heart looping defects, Isl1 −/− embryos show hypoplasia or complete absence of the right ventricle, the outflow tract and parts of the atria including the inflow tract [6]. Early in embryogenesis, Isl1 positive cells, which exhibit multipotent properties [4, 36], contribute to both the arterial (outflow tract) and the venous pole (SAN) of the primary heart tube. Once these progenitor cells differentiate into cardiomyocytes, Isl1 expression becomes downregulated and almost extinguished from E12.5 to E18.5 [27]. Small numbers of Isl1 positive cells can still be detected in the adult hearts, including the SAN [14, 24, 54].
Although signaling pathways up- and downstream of Isl1 have been investigated in the past [1, 6, 8, 26, 39], little is known about direct regulators controlling the transcriptional activity of this gene in cardiac cells. The only proteins known to regulate Isl1 expression so far via direct binding are ß-Catenin [28], Forkhead- and Gata4 transcription factors [22, 23] and the POU homeodomain transcription factor Oct1 [31]. While the binding sites for these regulators reside in promoter and enhancer regions up- or downstream of the gene, we could identify a regulatory element within an intronic region of ISL1.
In the present study, a connection between the homeodomain transcription factor Shox2 and Isl1 was revealed during cardiac development. We could show that Shox2 is essential for the expression of Isl1 in the SAN. Based on luciferase reporter studies and EMSA, our data also demonstrated that SHOX2 directly regulates ISL1 expression and that this regulation is conducted via the binding to an intronic regulatory element of ISL1. To study the transcriptional regulation of isl1 by shox2 in vivo, zebrafish was employed as a model. We have shown previously that shox2 loss of function in zebrafish embryos results in severe cardiac dysfunction with sinus bradycardia and intermittent sinus exit block after 72 h of development [2]. Similar data have also been published for Isl1 [9] and we could confirm these findings experimentally. Furthermore, it has been shown that both genes are expressed at the venous pole of the atrium in zebrafish embryos (48–72 hpf) [2, 57]. In the present study, we demonstrate a functional link between shox2 and isl1 in zebrafish embryos by partially rescuing the shox2-mediated bradycardia via ectopic expression of isl1. Previously, two separate phases of cardiomyocyte differentiation have been shown to exist in zebrafish. The first process, which regulates the differentiation of the venous pole comprising the sinus venosus requires Isl1 expression, while in a second Fgf8-dependent process new cells are added to the arterial pole of the heart [9]. Interestingly, in both Isl1 mutant fish and Shox2 knockout mice, Bmp4 expression is lost in the inflow tract (sinus venosus), but not the outflow tract of the developing heart [9, 42]. In addition, deficiency of both Isl1 and Shox2 in fish, as well as Shox2 deficiency in the mouse results in a bradycardia phenotype [2, 9]. The similarity of the phenotypes observed in Isl1 and Shox2 mutants point to a genetic link between these two genes in the development or function of the cardiac pacemaker. Taken together, our findings revealed an interesting interplay between Shox2 and Isl1 in the SAN, which contributes to a novel transcriptional regulation in the cardiac pacemaker system (Fig. 5).

Schematic representation of the epistatic relationship between Shox2 and Isl1. Schematic illustration of the mammalian conduction system highlighting a novel transcriptional regulation in the sinoatrial node. Previously we could show that Tbx5 regulates Shox2 in the SAN of the developing heart (12). In the present study, we demonstrate a novel epistatic relationship and a direct functional link between Shox2 and Isl1 in this compartment of the heart with important function in the pacemaking system. AVN atrioventricular node, HB bundle of His, ICV inferior caval vein, LA left atrium, LBB left bundle branch, LV left ventricle, PF Purkinje fibers, RA right atrium, RBB right bundle branch, RV right ventricle, SAN sinoatrial node, SCV superior caval vein
Based on the established functional data, SHOX2 as well as SHOX2 targets represent excellent candidates for human arrhythmias. While genetic variation in ISL1 is associated with congenital heart disease and cardiomyopathies [13, 47], SHOX2 has not been linked to any human phenotype yet. In a SHOX/Shox2 replacement mouse model, it was recently shown that a hypomorphic SHOX allele is not able to completely rescue the Shox2 −/− cardiac phenotype. Although the mice survive, they still show arrhythmias, suggesting that SHOX2 haploinsufficiency may lead to arrhythmia in humans [29]. Thus, it will be interesting to see if both SHOX2 and ISL1 are involved in human heart disease, especially arrhythmias.
A possible connection between Isl1 expressing second heart field-derived cells and the formation of the cardiac conduction system has been previously proposed [35, 48, 54]. As Shox2 directly regulates Isl1 expression specifically in the embryonic SAN, our data show for the first time a direct functional consequence for Isl1 in the pacemaking system of the developing heart. Taken together, our data add another piece to the puzzle of regulatory transcriptional networks controlling the development of the pacemaker.
References
Black BL (2007) Transcriptional pathways in second heart field development. Semin Cell Dev Biol 18:67–76. doi:10.1016/j.semcdb.2007.01.001
Blaschke RJ, Hahurij ND, Kuijper S, Just S, Wisse LJ, Deissler K, Maxelon T, Anastassiadis K, Spitzer J, Hardt SE, Schöler H, Feitsma H, Rottbauer W, Blum M, Meijlink F, Rappold G, Gittenberger-de Groot AC (2007) Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation 115:1830–1838. doi:10.1161/CIRCULATIONAHA.106.637819
Blaschke RJ, Monaghan AP, Schiller S, Schechinger B, Rao E, Padilla-Nash H, Ried T, Rappold G (1998) SHOT, a SHOX-related homeobox gene, is implicated in craniofacial, brain, heart, and limb development. Proc Natl Acad Sci USA 95:2406–2411. doi:10.1073/pnas.95.5.2406
Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR (2009) Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 460:113–117. doi:10.1038/nature08191
Buckingham M, Meilhac S, Zaffran S (2005) Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 6:826–835. doi:10.1038/nrg1710
Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S (2003) Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell 5:877–889. doi:10.3410/f.1016943.201654
Christoffels VM, Smits GJ, Kispert A, Moorman AF (2010) Development of the pacemaker tissues of the heart. Circ Res 106:240–254. doi:10.1161/CIRCRESAHA.109.205419
Cohen ED, Wang Z, Lepore JJ, Lu MM, Taketo MM, Epstein DJ, Morrisey EE (2007) Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J Clin Invest 117:1794–1804. doi:10.1172/JCI31731
de Pater E, Clijsters L, Marques SR, Lin Y-F, Garavito-Aguilar ZV, Yelon D, Bakkers J (2009) Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development 136:1633–1641. doi:10.1242/dev.030924
Dobrzynski H, Boyett MR, Anderson RH (2007) New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115:1921–1932. doi:10.1161/CIRCULATIONAHA.106.616011
Espinoza-Lewis RA, Liu H, Sun C, Chen C, Jiao K, Chen Y (2011) Ectopic expression of Nkx2.5 suppresses the formation of the sinoatrial node in mice. Dev Biol 356:359–369. doi:10.1016/j.ydbio.2011.05.663
Espinoza-Lewis RA, Yu L, He F, Liu H, Tang R, Shi J, Sun X, Martin JF, Wang D, Yang J, Chen Y (2009) Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nk2–5. Dev Biol 327:376–385. doi:10.1016/j.ydbio.2008.12.028
Friedrich FW, Dilanian G, Khattar P, Juhr D, Gueneau L, Charron P, Fressart V, Vilquin JT, Isnard R, Gouya L, Richard P, Hammoudi N, Komajda M, Bonne G, Eschenhagen T, Dubourg O, Villard E, Carrier L (2012) A novel genetic variant in the transcription factor Islet-1 exerts gain of function on myocyte enhancer factor 2C promoter activity. Eur J Heart Fail. doi:10.1093/eurjhf/hfs178
Genead R, Danielsson C, Andersson AB, Corbascio M, Franco-Cereceda A, Sylven C, Grinnemo KH (2010) Islet-1 cells are cardiac progenitors present during the entire lifespan: from the embryonic stage to adulthood. Stem Cells Dev 19:1601–1615. doi:10.1089/scd.2009.0483
Gittenberger-de Groot AC, Mahtab EAF, Hahurij ND, Wisse LJ, Deruiter MC, Wijffels MCEF, Poelmann RE (2007) Nkx2.5-negative myocardium of the posterior heart field and its correlation with Podoplanin expression in cells from the developing cardiac pacemaking and conduction system. Heart 122:115–122. doi:10.1002/ar.20406
Harland RM (1991) In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol 36:685–695. doi:10.1016/S0091-679X(08)60307-6
Hatcher CJ, Basson CT (2009) Specification of the cardiac conduction system by transcription factors. Circ Res 105:620–630. doi:10.1161/CIRCRESAHA.109.204123
Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ (2003) Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn 228:30–40. doi:10.1002/dvdy.10356
Jongbloed MR, Mahtab EA, Blom NA, Schalij MJ, Gittenberger-de Groot AC (2008) Development of the cardiac conduction system and the possible relation to predilection sites of arrhythmogenesis. Sci World J 8:239–269. doi:10.1100/tsw.2008.40
Just S, Berger IM, Meder B, Backs J, Keller A, Marquart S, Frese K, Patzel E, Rauch GJ, Katus HA, Rottbauer W (2011) Protein kinase D2 controls cardiac valve formation in zebrafish by regulating histone deacetylase 5 activity. Circulation 124:324–334. doi:10.1161/CIRCULATIONAHA.110.003301
Just S, Meder B, Berger IM, Etard C, Trano N, Patzel E, Hassel D, Marquart S, Dahme T, Vogel B, Fishman MC, Katus HA, Strahle U, Rottbauer W (2011) The myosin-interacting protein SMYD1 is essential for sarcomere organization. J Cell Sci 124:3127–3136. doi:10.1242/jcs.084772
Kang J, Nathan E, Xu SM, Tzahor E, Black BL (2009) Isl1 is a direct transcriptional target of Forkhead transcription factors in second-heart-field-derived mesoderm. Dev Biol 334:513–522. doi:10.1016/j.ydbio.2009.06.041
Kappen C, Salbaum JM (2009) Identification of regulatory elements in the Isl1 gene locus. Int J Dev Biol 53:935–946. doi:10.1387/ijdb.082819ck
Khattar P, Friedrich FW, Bonne G, Carrier L, Eschenhagen T, Evans SM, Schwartz K, Fiszman MY, Vilquin J-T (2011) Distinction between two populations of islet-1-positive cells in hearts of different murine strains. Stem Cells Dev 20:1043–1052. doi:10.1089/scd.2010.0374
Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld HH, Rotering H, Fortmueller L, Laakmann S, Verheule S, Schotten U, Fabritz L, Brown NA (2011) PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet 4:123–133. doi:10.1161/CIRCGENETICS.110.958058
Kwon C, Qian L, Cheng P, Nigam V, Arnold J, Srivastava D (2009) A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol 11:951–957. doi:10.1038/ncb1906
Laugwitz K-L, Moretti A, Caron L, Nakano A, Chien KR (2008) Islet1 cardiovascular progenitors: a single source for heart lineages? Development 135:193–205. doi:10.1242/dev.001883
Lin L, Cui L, Zhou W, Dufort D, Zhang X, Cai CL, Bu L, Yang L, Martin J, Kemler R, Rosenfeld MG, Chen J, Evans SM (2007) Beta-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc Natl Acad Sci USA 104:9313–9318. doi:10.1073/pnas.0700923104
Liu H, Chen CH, Espinoza-Lewis RA, Jiao Z, Sheu I, Hu X, Lin M, Zhang Y, Chen Y (2011) Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J Biol Chem 286:17029–17038. doi:10.1074/jbc.M111.234252
Liu H, Espinoza-Lewis RA, Chen C, Hu X, Zhang Y, Chen Y (2012) The role of Shox2 in SAN development and function. Pediatr Cardiol 33:882–889. doi:10.1007/s00246-012-0179-x
Liu Y, Li Y, Li T, Lu H, Jia Z, Wang W, Chen P, Ma K, Zhou C (2010) POU homeodomain protein OCT1 modulates islet 1 expression during cardiac differentiation of P19CL6 cells. Cell Mol Life Sci 68:1969–1982. doi:10.1007/s00018-010-0544-y
Loots GG (2008) Genomic identification of regulatory elements by evolutionary sequence comparison and functional analysis. Adv Genet 61:269–293. doi:10.1016/S0065-2660(07)00010-7
Mably JD, Mohideen MA, Burns CG, Chen JN, Fishman MC (2003) Heart of glass regulates the concentric growth of the heart in zebrafish. Curr Biol 13:2138–2147. doi:10.1016/j.cub.2003.11.055
Mangoni ME, Nargeot J (2008) Genesis and regulation of the heart automaticity. Physiol Rev 88:919–982. doi:10.1152/physrev.00018.2007
Mommersteeg MTM, Domínguez JN, Wiese C, Norden J, de Gier-de Vries C, Burch JBE, Kispert A, Brown NA, Moorman AFM, Christoffels VM (2010) The sinus venosus progenitors separate and diversify from the first and second heart fields early in development. Cardiovasc Res 87:92–101. doi:10.1093/cvr/cvq033
Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, Sun Y, Evans SM, Laugwitz K-L, Chien KR (2006) Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell 127:1151–1165. doi:10.1016/j.cell.2006.10.029
Munshi NV (2012) Gene regulatory networks in cardiac conduction system development. Circ Res 110:1525–1537. doi:10.1161/CIRCRESAHA.111.260026
Opthof T (1988) The mammalian sinoatrial node. Cardiovasc Drugs Ther 1:573–597. doi:10.1007/BF02125744
Park EJ, La Ogden, Talbot A, Evans S, Cai CL, Black BL, Frank DU, Moon AM (2006) Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development 133:2419–2433. doi:10.1242/dev.02367
Piperno G, Fuller MT (1985) Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. J Cell Biol 101:2085–2094. doi:10.1083/jcb.101.6.2085
Postma AV, Christoffels VM, Bezzina CR (2011) Developmental aspects of cardiac arrhythmogenesis. Cardiovasc Res 91:243–251. doi:10.1093/cvr/cvr134
Puskaric S, Schmitteckert S, Mori AD, Glaser A, Schneider KU, Bruneau BG, Blaschke RJ, Steinbeisser H, Rappold G (2010) Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart. Hum Mol Genet 19:4625–4633. doi:10.1093/hmg/ddq393
Rottbauer W, Just S, Wessels G, Trano N, Most P, Katus HA, Fishman MC (2005) VEGF-PLCgamma1 pathway controls cardiac contractility in the embryonic heart. Genes Dev 19:1624–1634. doi:10.1101/gad.1319405
Schrickel JW, Lickfett L, Lewalter T, Tiemann K, Nickenig G, Baba H, Heusch G, Schulz R, Levkau B (2012) Cardiomyocyte-specific deletion of survivin causes global cardiac conduction defects. Basic Res Cardiol 107:299. doi:10.1007/s00395-012-0299-8
Schweizer PA, Yampolsky P, Malik R, Thomas D, Zehelein J, Katus HA, Koenen M (2009) Transcription profiling of HCN-channel isotypes throughout mouse cardiac development. Basic Res Cardiol 104:621–629. doi:10.1007/s00395-009-0031-5
Srivastava D (2006) Making or breaking the heart: from lineage determination to morphogenesis. Cell 126:1037–1048. doi:10.1016/j.cell.2006.09.003
Stevens KN, Hakonarson H, Kim CE, Doevendans PA, Koeleman BP, Mital S, Raue J, Glessner JT, Coles JG, Moreno V, Granger A, Gruber SB, Gruber PJ (2010) Common variation in ISL1 confers genetic susceptibility for human congenital heart disease. PLoS ONE 5:e10855. doi:10.1371/journal.pone.0010855
Sun Y, Liang X, Najafi N, Cass M, Lin L, Cai CL, Chen J, Evans SM (2007) Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev Biol 304:286–296. doi:10.1016/j.ydbio.2006.12.048
Tessadori F, van Weerd JH, Burkhard SB, Verkerk AO, de Pater E, Boukens BJ, Vink A, Christoffels VM, Bakkers J (2012) Identification and functional characterization of cardiac pacemaker cells in zebrafish. PLoS ONE 7:e47644. doi:10.1371/journal.pone.0047644PONE-D-12-20487
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (2009) A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10:252–263. doi:10.1038/nrg2538
Vicente-Steijn R, Kolditz DP, Mahtab EA, Askar SF, Bax NA, Van Der Graaf LM, Wisse LJ, Passier R, Pijnappels DA, Schalij MJ, Poelmann RE, Gittenberger-de Groot AC, Jongbloed MR (2010) Electrical activation of sinus venosus myocardium and expression patterns of RhoA and Isl-1 in the chick embryo. J Cardiovasc Electrophysiol 21:1284–1292. doi:10.1111/j.1540-8167.2010.01790.x
Vincent D, Buckingham ME (2010) How to make a heart: the origin and regulation of cardiac progenitor cells. Curr Top Dev Biol 90:1–41. doi:10.1016/S0070-2153(10)90001-X
Wang J, Klysik E, Sood S, Johnson RL, Wehrens XH, Martin JF (2010) Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc Natl Acad Sci USA 107:9753–9758. doi:10.1073/pnas.0912585107
Weinberger F, Mehrkens D, Friedrich FW, Stubbendorff M, Hua X, Muller JC, Schrepfer S, Evans SM, Carrier L, Eschenhagen T (2012) Localization of Islet-1-positive cells in the healthy and infarcted adult murine heart. Circ Res 110:1303–1310. doi:10.1161/CIRCRESAHA.111.259630
Westerfield M (1993) The zebrafish book; a guide for the laboratory use of zebrafish (Brachydanio rerio). University of Oregon Press, Eugene
Wiese C, Grieskamp T, Airik R, Mommersteeg MT, Gardiwal A, de Gier-de Vries C, Schuster-Gossler K, Moorman AF, Kispert A, Christoffels VM (2009) Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ Res 104:388–397. doi:10.1161/CIRCRESAHA.108.187062
Witzel HR, Jungblut B, Choe CP, Crump JG, Braun T, Dobreva G (2012) The LIM protein Ajuba restricts the second heart field progenitor pool by regulating Isl1 activity. Dev Cell 23:58–70. doi:10.1016/j.devcel.2012.06.005
Wolf CM, Berul CI (2006) Inherited conduction system abnormalities—one group of diseases, many genes. J Cardiovasc Electrophysiol 17:446–455. doi:10.1111/j.1540-8167.2006.00427.x
Acknowledgments
We thank the Developmental Studies Hybridoma Bank (DSHB, University of Iowa) for the Isl1 antibody and Maria Saile for help with the microarray analyses. This work was supported by the Deutsche Forschungsgemeinschaft [RA 380/14-2]; the Bundesministerium für Bildung und Forschung [01GS1104 NGFNplus]; by the DZHK (Deutsches Zentrum für Herz-Kreislauf-Forschung—German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Hoffmann, S., Berger, I.M., Glaser, A. et al. Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res Cardiol 108, 339 (2013). https://doi.org/10.1007/s00395-013-0339-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-013-0339-z