
Abstract
Flos Lonicerae Japonicae (FLJ), the flower bud of Lonicera japonica Thunb. (Caprifoliaceae), is a widely used traditional Chinese medicine with various pharmacological activities. Luteoloside is a major active compound and a quality control marker of FLJ. Luteolin-7-O-glucuronide (LG), an analog of luteoloside, was conjugated with bovine serum albumin (BSA) and ovalbumin (OVA) to create the immunogen and coating antigen, respectively. A sensitive and specific monoclonal antibody (mAb), designated as mAb3A4, was generated with LG-BSA. To screen the authenticity and quality of FLJ, an indirect competitive enzyme-linked immunosorbent assay (icELISA) was established. The concentration of luteoloside producing 50 % inhibition and the working range of the icELISA were 42.3 and 9.1–258.1 μg L−1, respectively. The icELISA showed cross-reactivity values of 2414, 402, 230, and <1 % for LG, baicalin, scutellarin, and other analogs of luteoloside, respectively. The average recovery of luteoloside in the FLJ samples as determined by icELISA ranged from 83.0 to 112.5 %. The luteoloside content was determined for different Lonicera herbal samples with icELISA, and the results were confirmed by high-performance liquid chromatography analysis. Thus, this icELISA is suitable for the quality assurance of FLJ samples.

Specific monoclonal antibody-based enzyme-linked immunosorbent assay for luteoloside
Similar content being viewed by others

Introduction
Flos Lonicerae Japonicae (FLJ, Jin-Yin-Hua in Chinese), the flower bud of Lonicera japonica Thunb. (Caprifoliaceae), is one of the most famous traditional Chinese medicines (TCMs) and is officially listed in the Chinese Pharmacopoeia [1]. It has been used for many years in TCM for the treatment of various diseases, including fever, arthritis, and infections [2, 3]. There is a great demand for FLJ, especially during infectious disease outbreaks, such as SARS and flu, for its excellent anti-viral and anti-inflammatory effects [4–6]. Currently, it has commonly been added to beverages, health products, food, and cosmetics due to its proven health benefits and low toxicity [7].
In the monograph of the Chinese Pharmacopoeia (2005 edition; 2010 edition), Lonicera japonica Thunb., the most widespread and traditionally used species, was documented as the unique origin of FLJ [1, 8]. However, in a previous edition (2000 edition), the plant origin of FLJ also included Lonicera hypoglauca Miq., Lonicera confusa DC., and Lonicera dasystyla Rehd [9]. Recently, the former two species combined with Lonicera macranthoides Hand.-Mazz. and Lonicera fulvotomentosa Hsu et S.C. Cheng were classified as Flos Lonicerae (Shan-Yin-Hua in Chinese). The two Lonicera herbal drugs, Shan-Yin-Hua and FLJ, have been unintentionally mislabeled and confused on Chinese markets due to their complicated plant sources. However, different habitats, harvest times, drying processes, and extraction methods would result in different chemical constituents and a different quality of FLJ. Therefore, a reliable, simple, rapid, and high-throughput method is required to screen the authenticity and quality of FLJ on the market to guarantee safety and stable therapeutic effects.
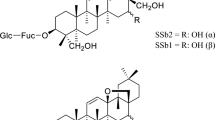
To date, more than 140 compounds including essential oils, flavones, organic acids, triterpenoid saponins, and iridoids have been isolated and identified from FLJ [7]. Before 2005, chlorogenic acid, one of the major active compounds in FLJ, served as its unique quality control marker. However, luteoloside (luteolin-7-O-glucoside, Fig. 1), another active compound from FLJ, and cynaroside were added as the second quality control markers in the monograph of the Chinese Pharmacopoeia (2005 edition), as there were no significant differences in chlorogenic acid contents among the Lonicera species mentioned. The luteoloside in FLJ may be partly responsible for its anti-bacterial and anti-oxidant activities [10, 11]. In addition, many other pharmacological activities, including anti-asthmatic, anti-hyperglycemic, anti-mutagenic, and anti-genotoxic effects, were discovered [12–16]. Luteoloside also exhibited beneficial effects on wound healing [17] and anti-cancer activity [18–21].

Chemical structure of luteolin-7-O-glucuronide (A) and luteoloside (B)
Numerous methods have been developed for the detection of luteoloside in FLJ, including high-performance liquid chromatography (HPLC) coupled with various detection systems (UV, DAD, and ELSD) [22, 23] or in tandem with mass spectrometry (MS) [24–26]. Though HPLC is the most widely utilized method for the analysis of natural herbs, several shortcomings including tedious sample preparation procedures, high solvent consumption, and short column lifetime (not simply due to numerous coexistent ingredients in herbs) make it unsuitable for rapid analysis. Compared to the instrumental methods above, enzyme-linked immunosorbent assay (ELISA) is simple, fast, highly specific, sensitive, inexpensive, and a promising high-throughput screening method for the quantitative and/or qualitative analysis of natural bioactive compounds [27, 28].
To our knowledge, immunoassays for luteoloside have not yet been reported. In the present work, a sensitive and selective monoclonal antibody (mAb) against luteoloside was produced and an indirect competitive ELISA (icELISA) was developed for the detection of luteoloside in Lonicera herbal samples. Furthermore, the ELISA was performed for the analysis of luteoloside and then verified by HPLC.
Materials and methods
Reagents and apparatus
Luteoloside (99 % purity), luteolin-7-O-glucuronide (LG, 99 % purity; Fig. 1), and the other analogs of luteoloside used for cross-reactivity (CR) studies were purchased from the National Institutes for Food and Drug Control (Beijing, China). Hypoxanthine, aminopterin, and thymidine (HAT); cell freezing medium-dimethyl sulfoxide (DMSO) (serum free); l-glutamine; streptomycin; penicillin; goat anti-mouse IgG conjugated with horseradish peroxidase (IgG-HRP); polyethylene glycol (PEG)-2000; N-hydroxysuccinimide (NHS); dicyclohexylcarbodiimide (DCC); N,N-dimethylformamide (DMF); bovine serum albumin (BSA); ovalbumin (OVA); complete and incomplete Freund’s adjuvant; and o-phenylenediamine (OPD) were purchased from Sigma (St. Louis, MO, USA). Cell culture media (Dulbecco’s modified Eagle’s medium, DMEM) and fetal bovine serum (FBS) were obtained from Thermo (Waltham, MA, USA). Chromatography-grade methanol and acetonitrile were obtained from Fisher Scientific (NJ, USA). All other chemicals used were of analytical grade.
Cell culture plates and 96-well polystyrene microtiter plates were purchased from Costar (Corning, NY, USA). An electric heating constant-temperature incubator (ZXDR-2800) was purchased from Shanghai Zhicheng Analytical Instrument Manufacturing Co., Ltd. (Shanghai, China). A direct-heat CO2 incubator (311), an automated plate washer (Wellwash 4 MK2), and a microplate reader (Multiskan FC) were purchased from Thermo. The Agilent 1200 HPLC system equipped with a quaternary pump, an autosampler, an online degasser, a thermostatically controlled column compartment, and an Agilent 1260 DAD detector was obtained from Agilent (Agilent Technologies, Santa Clara, CA, USA).
The HAT-sensitive Balb/c mouse myeloma cell line SP2/0-Ag14 used in the fusion experiment was obtained from ATCC (Manassas, VA, USA). Female Balb/c mice were purchased from the Laboratory Animal Center of the Institute of Genetics and Developmental Biology (Beijing, China). All the experiments were approved by the Animal Care Committee of China Agricultural University, and all efforts were made to minimize suffering.
Buffers and solutions
The buffers and solutions used here were the same as those previously reported [29]. They included coating buffer (0.05 M carbonate buffer, pH 9.6), phosphate-buffered saline (PBS) (0.01 M phosphate buffer containing 0.9 % NaCl, pH 7.5), PBS with 0.1 % (v/v) Tween-20 (PBST), PBST containing 0.5 % (w/v) gelatin (PBSTG), citrate-phosphate buffer (0.01 M citric acid and 0.03 M Na2HPO4, pH 5.5), substrate solution (4 μL of 30 % H2O2 added to 10 mL citrate-phosphate buffer containing 2 mg mL−1 OPD), and a stop solution (2 M H2SO4). Luteoloside standard solutions of various concentrations were prepared by diluting the stock solution (1 mg mL−1, made by dissolving luteoloside powder in DMF) with methanol.
Preparation of immunogen and coating antigen
LG, an analog of luteoloside, was used as the hapten to conjugate with the carrier protein. The active ester method was used to prepare the LG-BSA and LG-OVA conjugates as the immunogen and coating antigen, respectively [29]. DCC (7.5 mg) was added to a stirring mixture of LG (8.8 mg) and NHS (4.3 mg) dissolved in 2 mL DMF. The mixture was stirred overnight at 4 °C and then centrifuged. The supernatant was divided into two equal aliquots and added dropwise to 20 mg BSA or OVA in 2 mL of 0.05 M carbonate buffer (pH 9.6). The solution was stirred overnight at 4 °C. The reaction mixture was lyophilized and stored at −40 °C after dialysis against 0.01 M PBS (pH 7.5) for 3 days with two changes per day.
Immunization, mAb production, and characterization
The protocols of immunization, mAb production, and purification were similar to those previously described by Wang et al. [30]. Specifically, six female Balb/c mice (6–8 weeks old) were immunized subcutaneously with 0.1 mg of LG-BSA conjugate emulsified in Freund’s complete adjuvant. Two subsequent injections were carried out at 2-week intervals with Freund’s incomplete adjuvant. Five days after the third injections, the sera were tested for anti-luteoloside antibody titer and for luteoloside recognition properties in icELISA. The splenocytes of the mouse with the highest titer and best specificity were fused with the SP2/0 using PEG-2000. The plates were incubated at 37 °C in the CO2 incubator. Positive hybridomas were cloned by limiting dilution and expanded after screening by ELISA approximately 1 week after fusion. The clone with a high antibody titer and good sensitivity in the culture supernatant was expanded in mice for the production of mAb in ascites. The mAbs were purified by ammonium sulfate precipitation. The immunoglobulin isotype was determined with a mouse antibody isotyping kit (Pierce, Rockford, IL, USA).
icELISA
Microplate wells were coated with LG-OVA (1.0 μg mL−1, 100 μL per well in coating buffer) at 37 °C for 3 h. After four PBST washes, the plate was blocked with 100 μL per well of 3 % nonfat milk in PBS for 30 min at 37 °C and then washed with PBST four times. Various concentrations of the standard or samples in PBSTG (50 μL each) were added into each well, followed by the addition of 50 μL of sera, cell supernatant, or purified mAb solution diluted in PBSTG. The plate was incubated at 37 °C for 30 min and then washed again with PBST four times to remove any unbound antibodies. Goat anti-mouse IgG-HRP diluted in PBSTG (1.0 μg mL−1) was added (100 μL per well). After incubation at 37 °C for 30 min, the plate was washed again four times with PBST. Substrate solution (100 μL) was pipetted into each well. The reaction was terminated by adding 50 μL of 2 M H2SO4 per well. The absorbance was measured at 492 nm with the Multiskan FC microplate reader. The detection data of each analyte was calculated using Origin Pro 8.0 software (Origin Lab, USA).
Assay specificity
Assay specificity was evaluated by cross-reactivity (CR) with a set of structural analogs of luteoloside. The CR of luteoloside was calculated according to the formula:
Sample preparation and extraction
Lonicera herbal samples including FLJ and Shan-Yin-Hua were collected from different cultivation regions of China. The extraction procedures followed the regulations of the Chinese Pharmacopoeia (2010 edition) [1]. Namely, the samples were powdered and dried at 60 °C in an oven for 3 h. The pulverized plant samples (2.0 g) were accurately weighed and suspended in 50 mL of 70 % aqueous ethanol (v/v), ultrasonically extracted for 1 h, and then cooled at room temperature. Additional 70 % aqueous ethanol was added to compensate for the weight lost. The extracts were centrifuged at 8000g for 10 min. The supernatants were collected and divided into two equal aliquots, and then evaporated for use. One resultant residue was dissolved in 20 mL of 70 % aqueous ethanol and detected in the icELISA after dilution with PBS. The dilution rate was 500 and 100 for FLJ and Shan-Yin-Hua, respectively. The other resultant residue was dissolved in 10 mL of 70 % aqueous ethanol, and filtered through a 0.22 μm Millipore membrane for HPLC analysis.
Assay recovery experiment
Powdered samples of FLJ (100 mg), the luteoloside contents of which were quantified by icELISA, were spiked with luteoloside at concentrations of 0.1, 0.2, 0.4, 0.6, and 0.8 mg g−1. After storage at 4 °C overnight, the samples were extracted according to the extraction procedure for ELISA as described in the previous section.
HPLC analysis
A Zorbax SB-phenyl column (4.6 × 250 mm, 5 μm) (Agilent Technologies) was used to separate luteoloside. The column temperature was maintained at 35 °C. A mixture of solvent A (0.4 % v/v acetic acid aqueous) and solvent B (acetonitrile) was used as the mobile phase at a flow rate of 1.0 mL min−1. The gradient elution program was 0–15 min, 10–20 % B; 15–30 min, 20 % B; and 30–40 min, 20–30 % B. The DAD detector was set to monitor at 350 nm. The sample volume injected was 10 μL. All data were collected and analyzed by Agilent ChemStation.
Results and discussion
Preparation of protein-hapten conjugates
Luteoloside itself has an o-hydroxy moiety in the glucosyl group that can be used for conjugation with a carrier protein via periodate oxidation [31]. The reaction product is unstable if the reductant is added improperly. Furthermore, the phenolic hydroxyl group may be oxidized if excessive NaIO4 is used or the reaction time is too long. The LG molecule possesses a carboxylic acid group in a position different from that of luteoloside, which makes it easy to covalently link it directly to carrier proteins via the active ester method. The concentrations of hapten bound to the carrier proteins were estimated by UV-VIS as described previously [32]. The results indicated that the hapten was successfully coupled with the carrier proteins. The molar ratios of LG to the proteins were estimated to be 10:1 and 8:1 for LG-BSA and LG-OVA, respectively.
Production and characterization of mAbs
The clone, named mAb3A4, with the best sensitivity and selectivity was expanded for ascites production. The titer (the maximum serum dilution that gave an absorbance of 1.0 at noncompetitive assay conditions) of the ascites was approximately 2 × 104. The dissociation constant (K d) of mAb3A4, 2.8 × 10−10 M, was determined using the method of Beatty [33]. mAb3A4 is an IgG1 isotype that has κ light chains.
Optimization of icELISA conditions
The optimal concentrations of coating antigen, mAb, and goat anti-mouse IgG-HRP were screened by checkerboard titration. Concentrations of 1.0 μg mL−1 LG-OVA, 1.0 μg mL−1 mAb3A4, and 0.1 μg mL−1 goat anti-mouse IgG-HRP were selected and used throughout this work. Under these optimal assay conditions, a standard curve was plotted with logarithmic concentrations of luteoloside (400, 200, 100, 50, 25, 12.5, 6.25, 3.13, 1.56, and 0 μg L−1) as the lateral coordinates and the corresponding B/B 0 values as the longitudinal coordinates. As shown in Fig. 2, the representative inhibition curve for luteoloside by icELISA was between 9.1 and 258.1 μg L−1 (based on 20–80 % binding inhibition of mAb3A4 to luteoloside), with an IC50 value of approximately 42.3 μg L−1.

Standard inhibition curve of luteoloside by icELISA, obtained under optimized conditions. B 0 and B were absorbance in the absence and presence of competitors, respectively. Each value represents the mean of three replicates
Assay specificity
The CR values of the anti-luteoloside mAb against the structurally related compounds were checked to evaluate the icELISA assay specificity. As indicated in Table 1, the mAb showed a negligible CR value to most of the compounds except LG, baicalin, and scutellarin, as they all have a very similar chemical structure. The CR values of mAb3A4 with LG, baicalin, and scutellarin were 2414, 402, and 230 %, respectively. Compared to the chemical structure of luteoloside, it is suggested that the glucosides and the carboxyl group at the C5″ position may play a key role in recognition by mAb3A4. Moreover, the mAb had only a slight CR value with apigenin, luteolin, baicalein, and wogonin, and no inhibition was observed at up to 40,000 μg L−1 of the other six compounds. Until now, approximately 30 flavones have been isolated from FLJ [7]. LG, baicalin, and scutellarin had not been detected or reported in FLJ, though the mAb showed high CR values with them.
Assay recovery experiment
The recovery rates of each spiked luteoloside were calculated using the spiked and recovered amounts of luteoloside in the same concentration range. The mean recovery of luteoloside in the spiked samples ranged from 83.0 to 112.5 % with low RSD values (1.1–6.7 %), as indicated in Table 2.
Correlation of luteoloside contents in Lonicera herbal samples, as determined by ELISA and HPLC
The icELISA was applied to determine the content of luteoloside in Lonicera herbal samples (Table 3). The luteoloside contents in the samples were also analyzed by HPLC. The results obtained by icELISA agreed well with those from HPLC. The linear regression equation between the two methods was Y = 0.912X + 0.023, with a correlation of 0.99786 (Fig. 3).

Correlation between luteoloside content of Lonicera herbal samples determined by ELISA and by HPLC
As shown in Table 3, the results determined by icELISA were similar to those detected by HPLC. Furthermore, the luteoloside contents in FLJ were higher than in Shan-Yin-Hua. More specifically, the luteoloside contents in the FLJ samples varied from 450 to 1037 μg g−1, but the two samples of Shan-Yin-Hua from Beijing and Guizhou were lower than 200 μg g−1. As regulated by the Chinese Pharmacopoeia (2010 edition; luteoloside content should be no less than 0.05 %), most of the FLJ origins for L. japonica Thunb. were qualified, except the sample from Anhui, whereas the samples of Shan-Yin-Hua were not up to these standards. The results indicated that luteoloside was reasonable as a quality marker and that the developed icELISA method was useful in screening the authenticity and quality of FLJ.
Conclusions
To our knowledge, this is the first mAb against luteoloside produced and applied to an icELISA for the determination of luteoloside in FLJ samples. The results obtained from the icELISA corroborated those from the HPLC analysis. Thus, icELISA was a simple, rapid, high-throughput, cost-effective, and reliable method compared with other instrumental analyses. Moreover, icELISA could be used as a quality control method for FLJ. The luteoloside content varied remarkably in different Lonicera herbal samples. However, L. japonica Thunb. has a significantly higher luteoloside content than Shan-Yin-Hua.
References
National Committee of Pharmacopoeia. Pharmacopoeia of the People’s Republic of China, vol. 1. Beijing: Chemical Industry Press; 2010. p. 205.
Ma SC, Du J, But PPH, Deng XL, Zhang YW, Ooi VEC, et al. Antiviral Chinese medicinal herbs against respiratory syncytial virus. J Ethnopharmacol. 2002;79:205–11.
Kang M, Jung I, Hur J, Kim SH, Lee JH, Kang JY, et al. The analgesic and anti-inflammatory effect of WIN-34B, a new herbal formula for osteoarthritis composed of Lonicera japonica Thunb and Anemarrhena asphodeloides BUNGE in vivo. J Ethnopharmacol. 2010;131:485–96.
Jiang M, Han YQ, Zhou MG, Zhao HZ, XIAO X, HOU YY, et al. The screening research of anti-inflammatory bioactive markers from different flowering phases of Flos Lonicerae Japonicae. PLoS ONE. 2014;9, e96214.
Yoo HJ, Kang HJ, Song YS, Park EH, Lim CJ. Anti-angiogenic, antinociceptive and anti-inflammatory activities of Lonicera japonica extract. J Pharm Pharmacol. 2008;60:779–86.
Cheng BC, Ma XQ, Kwan HY, Tse KW, Cao HH, Su T, et al. A herbal formula consisting of Rosae Multiflorae Fructus and Lonicerae Japonicae Flos inhibits inflammatory mediators in LPS-stimulated RAW 264.7 macrophages. J Ethnopharmacol. 2014;153:922–7.
Shang X, Pan H, Li M, Miao X, Ding H. Lonicera japonica Thunb.: ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J Ethnopharmacol. 2011;138:1–21.
National Committee of Pharmacopoeia. Pharmacopoeia of the People’s Republic of China, vol. 1. Beijing: Chemical Industry Press; 2005. p. 152.
National Committee of Pharmacopoeia. Pharmacopoeia of the People’s Republic of China, vol. 1. Beijing: Chemical Industry Press; 2000. p. 77.
Xiong J, Li S, Wang W, Hong Y, Tang K, Luo Q. Screening and identification of the antibacterial bioactive compounds from Lonicera japonica Thunb. leaves. Food Chem. 2013;138:327–33.
Tang D, Li HJ, Chen J, Guo CW, Li P. Rapid and simple method for screening of natural antioxidants from Chinese herb Flos Lonicerae Japonicae by DPPH-HPLC-DAD-TOF/MS. J Sep Sci. 2008;31:3519–26.
Zhu X, Zhang H, Lo R. Phenolic compounds from the leaf extract of artichoke (Cynara scolymus L.) and their antimicrobial activities. J Agric Food Chem. 2004;52:7272–8.
Ooi LS, Wang H, He Z, Ooi VE. Antiviral activities of purified compounds from Youngia japonica (L.) DC (Asteraceae, Compositae). J Ethnopharmacol. 2006;106:187–91.
Jin M, Yang JH, Lee E, Lu Y, Kwon S, Son KH, et al. Antiasthmatic activity of luteolin-7-O-glucoside from ailanthus altissima through the down regulation of T helper 2 cytokine expression and inhibition of prostaglandin E2 production in an ovalbumin-induced asthma model. Biol Pharm Bull. 2009;32:1500–3.
Choi JS, Young HS, Kim BW. Hypoglycemic and hypolipemic effects of Ixeris dentata in diabetic rats. Arch Pharm Res. 1990;13:269–73.
Boldbaatar D, El-Seedi HR, Findakly M, Jabri S, Javzan B, Choidash B, et al. Antigenotoxic and antioxidant effects of the Mongolian medicinal plant Leptopyrum fumarioides (L): an in vitro study. J Ethnopharmacol. 2014;155:599–606.
Suntar I, Kupeli Akkol E, Keles H, Yesilada E, Sarker SD, Arroo R, et al. Efficacy of Daphne oleoides subsp. kurdica used for wound healing: identification of active compounds through bioassay guided isolation technique. J Ethnopharmacol. 2012;141:1058–70.
Baskar AA, Ignacimuthu S, Michael GP, Al Numair KS. Cancer chemopreventive potential of luteolin-7-O-glucoside isolated from Ophiorrhiza mungos Linn. Nutr Cancer. 2011;63:130–8.
Sun X, Sun GB, Wang M, Xiao J, Sun XB. Protective effects of cynaroside against H(2)O(2)-induced apoptosis in H9c2 cardiomyoblasts. J Cell Biochem. 2011;112:2019–29.
Hwang YJ, Lee EJ, Kim HR, Hwang KA. Molecular mechanisms of luteolin-7-O-glucoside-induced growth inhibition on human liver cancer cells: G2/M cell cycle arrest and caspase-independent apoptotic signaling pathways. BMB Rep. 2013;46:611–6.
Fan SH, Wang YY, Lu J, Zheng YL, Wu DM, Li MQ, et al. Luteoloside suppresses proliferation and metastasis of hepatocellular carcinoma cells by inhibition of NLRP3 inflammasome. PLoS ONE. 2014;9, e89961.
Nalewajko-Sieliwoniuk E, Malejko J, Mozolewska M, Wolyniec E, Nazaruk J. Determination of polyphenolic compounds in Cirsium palustre (L.) extracts by high performance liquid chromatography with chemiluminescence detection. Talanta. 2015;133:38–44.
Chen CY, Qi LW, Li HJ, Li P, Yi L, Ma HL, et al. Simultaneous determination of iridoids, phenolic acids, flavonoids, and saponins in Flos Lonicerae and Flos Lonicerae Japonicae by HPLC-DAD-ELSD coupled with principal component analysis. J Sep Sci. 2007;30:3181–92.
Li R, Liu SK, Song W, Wang Y, Li YJ, Qiao X, et al. Chemical analysis of the Tibetan herbal medicine Carduus acanthoides by UPLC/DAD/qTOF-MS and simultaneous determination of nine major compounds. Anal Methods. 2014;6:7181–9.
Ren MT, Chen J, Song Y, Sheng LS, Li P, Qi LW. Identification and quantification of 32 bioactive compounds in Lonicera species by high performance liquid chromatography coupled with time-of-flight mass spectrometry. J Pharm Biomed Anal. 2008;48:1351–60.
Yin R, Han F, Tang Z, Liu R, Zhao X, Chen X, et al. UFLC-MS/MS method for simultaneous determination of luteolin-7-O-gentiobioside, luteolin-7-O-beta-D-glucoside and luteolin-7-O-beta-D-glucuronide in beagle dog plasma and its application to a pharmacokinetic study after administration of traditional Chinese medicinal preparation: Kudiezi injection. J Pharm Biomed Anal. 2013;72:127–33.
He SP, Tan GY, Li G, Tan WM, Nan TG, MinWang B, et al. Development of a sensitive monoclonal antibody-based enzyme-linked immunosorbent assay for the antimalaria active ingredient artemisinin in the Chinese herb Artemisia annua L. Anal Bioanal Chem. 2009;393:1297–303.
Kido K, Morinaga O, Shoyama Y, Tanaka H. Quick analysis of baicalin in Scutellariae Radix by enzyme-linked immunosorbent assay using a monoclonal antibody. Talanta. 2008;77:346–50.
Zhao J, Li G, Wang BM, Liu W, Nan TG. Development of a monoclonal antibody-based enzyme-linked immunosorbent assay for the analysis of glycyrrhizic acid. Anal Bioanal Chem. 2006;386:1735–40.
Wang Y, Yang H, Pschenitza M, Niessner R, Li Y, Knopp D, et al. Highly sensitive and specific determination of mercury(II) ion in water, food and cosmetic samples with an ELISA based on a novel monoclonal antibody. Anal Bioanal Chem. 2012;403:2519–28.
Nan TG, Wu SQ, Zhao HW, Tan WM, Li ZH, Zhang QC, et al. Development of a secondary antibody thio-functionalized microcantilever immunosensor and an ELISA for measuring ginsenoside Re content in the herb ginseng. Anal Chem. 2012;84:4327–33.
Yuan Y, Hua X, Li M, Yin W, Shi H, Wang M. Development of a sensitive indirect competitive enzyme-linked immunosorbent assay based on the monoclonal antibody for the detection of benzothiostrobin residue. RSC Adv. 2014;4:24406.
Beatty JD, Beatty BG, Vlahos WG. Measurement of monoclonal antibody affinity by non-competitive enzyme immunoassay. J Immunol Methods. 1987;100:173–9.
Acknowledgments
The work was supported by the specific funds of the Traditional Chinese Medicine industry (No. 201407003) and the National Natural Science Foundation of China (No. 81373959). The authors would like to thank Professor Qing X. Li from the Department of Molecular Biosciences and Bioengineering of the University of Hawaii for careful review of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
This study was performed in strict accordance with the standards described in the “Guide for the Care and Use of Laboratory Animals” (National Research Council Commission on Life Sciences, 1996 edition). All the animal treatment procedures were approved by the Animal Care Committee of China Agricultural University (CAU), and all efforts were made to minimize suffering. The mice were housed under controlled temperature (22 ± 2 °C) and lighting (12-h light/12-h darkness) with food and water ad libitum in air-conditioned rooms. All the experimental mice were killed by cervical dislocation.
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Published in the topical collection Immunoanalysis for Environmental Monitoring and Human Health with guest editors Shirley J. Gee, Ivan R. Kennedy, Alice Lee, Hideo Ohkawa, Tippawan Prapamontol, and Ting Xu.
Bo Zhang and Tiegui Nan contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhang, B., Nan, T., Zhan, Z. et al. Development of a monoclonal antibody-based enzyme-linked immunosorbent assay for luteoloside detection in Flos Lonicerae Japonicae. Anal Bioanal Chem 408, 6053–6061 (2016). https://doi.org/10.1007/s00216-016-9396-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9396-0