
Abstract
Size-exclusion chromatography (SEC) and capillary zone electrophoresis (CZE) coupled with inductively coupled plasma mass spectrometry were applied to characterize low, medium, and high molecular weight cadmium complexes with glutathione and phytochelatins (PCs). The dominant stoichiometry of the complexes formed in vitro was established as 1:1 using electrospray ionization mass spectrometry. Calculated molecular masses of Cd1L1 complexes were used for calibration of the SEC and CZE methods. The results showed a lower (2 kDa) SEC column exclusion limit for cadmium complexes compared with free peptides (10 kDa), and most of the high molecular weight cadmium species were eluted in the void volume of the column. Moreover, the CZE method based on the semiempirical model of Offord to elucidate peptide migration allowed us to show a high propensity of Cd–PC complexes for polymorphism on complexation, which was also observed for extracts of Arabidopsis thaliana treated with cadmium. All the information presented is vital for understanding the mechanism of metal deactivation in plants.

Estimation of molecular mass of Cd-thiopeptide complexes by size electrophoretic mobility
Similar content being viewed by others


Introduction
Phytochelatins (PCs) are thiopeptides of the general formula (γ-GluCys) n Gly, where n usually ranges from 2 to 5 [1]. Their main function is to bind metal ions through the thiol groups [2]; therefore, PCs play a key role in detoxification and homeostasis of metal and metalloid ions in plants, especially for Cd2+, Hg2+, Ag+, Sb3+ [3], and species of arsenic and selenium, leading to the development of the new field in plant biochemistry of metallothiolomics [4]. The enzymatic synthesis of PCs is activated by binding metal ions to PC synthase. Recent findings confirm the postulate that complexes of a metal with glutathione (GSH) or PCs can be a suitable cosubstrate to create a metal–enzyme complex through ligand exchange [2, 5]. In consequence, PCs are expected to play two contradictory roles: (1) as ligands creating reversible complexes for activation of PC synthase and (2) as ligands creating stable complexes responsible for deactivation of heavy metals and termination of their synthesis [2]. As the stability of metal complexes increases with the length of PCs, the synthesis of long-chain PCs should be favored [5, 6]. However, PC5 is usually the longest owing to high and hazardous consumption of GSH [1]. Mutoh and Hayashi [7] reported that metal complexes of PCs can rearrange themselves into agglomerates with higher efficiency of cadmium binding. They stated that the ability of plants to create high molecular weight (HMW) complexes through medium molecular weight (MMW) complexes from low molecular weight (LMW) complexes is more productive for detoxification of toxic metals than for synthesis of longer PCs [7]. HMW complexes created by cadmium ions were observed mainly by using size-exclusion chromatography (SEC) with inductively coupled plasma (ICP) mass spectrometry (MS) for detection [8, 9]. Lately, Bräutigam et al. [10] confirmed this theory indirectly by identification of different PC conjugates with disulfide bridges instead of long-chain PCs in algal extracts. Still, only the occurrence of LMW complexes was confirmed by electrospray ionization (ESI) MS, probably owing to low efficiency of the ionization process [11].
Even though PCs are widely distributed in the plant kingdom, little information about the stoichiometries and kinetics of the complexes formed between the various PC oligomers and metal ions has been obtained. In spite of many analytical problems related to the fairly low stability of metal–peptide complexes and even of PCs themselves (because of oxidation through the creation of disulfide bridges induced by the presence of the metal) [10, 12], many attempts to characterize GSH and PC complexes by different techniques have been undertaken. The first results were obtained by nano-ESI MS for plant extracts [13]. Spectrometric signals corresponding to the cadmium complexes with longer PCs were observed only after partial decomposition of the complexes in acidic solution used as the mobile phase during separation by reversed-phase liquid chromatography [13].
SEC coupled with ICP MS was proposed as the most suitable technique because the complexes are separated via gentle steric interactions and it offers the possibility to characterize compounds by their size. HMW complexes extracted from different plants species (Silene vulgaris and Arabidopsis thaliana) and their formation in vitro from a mixture of PC standards with cadmium ions were observed in the chromatograms [8, 9]. However, the negative residual charges on the stationary phase were responsible for strong interactions with cadmium ions, leading to low recovery of metal (from 50 to 60 %). The addition of sodium chloride to the mobile phase increased the chromatographic recovery of cadmium up to 80 % owing to the “covering effect” [9]. The same effect can be obtained taking into consideration active silanol groups by addition of tetrabutylammonium hydroxide or another amine used as an ion-pairing agent in the case of reserved-phase chromatography. The recovery of cadmium can even reach 90 % [14]. However, only LMW complexes were observed.
ESI MS was also used for the characterization of Cd–GSH and Cd–PC complexes from standard mixtures injected directly into the ESI source [11, 12, 15] or after separation by SEC using a mobile phase containing ammonium acetate or ammonium carbonate, with the postcolumn addition of methanol [11, 12]. It is important to note that despite different buffers being applied, similar cadmium complexes were found. However, the proposed stoichiometry was limited to LMW complexes such as Cd1L1 and Cd1L2 for GSH and PC2, and Cd1L1, Cd2L1, and Cd3L1 for PC2, PC3, and PC4. Other approaches used were also limited to LMW cadmium complexes of GSH and PCs, such as potentiometric pH titration [16], UV spectroscopy [17], NMR spectroscopy [18], and differential pulse voltammetry/polarography [15, 16, 19].
Taking into consideration the ambiguous composition and often inadequate stability of the complexes investigated as well as the problems related to the limitations of each technique (e.g., selectivity, sensitivity, and incompatibility with physiological conditions), all the results should be confirmed by an alternative method. Capillary zone electrophoresis (CZE) is capable of providing useful information required for the characterization of unstable metal–bioligand complexes because of limited interactions with the capillary surface. In such a technique, being based on monitoring the electrophoretic mobility (m ef) of an analyte in a well-defined chemical environment, CZE provides data reflecting the mass-to-charge ratio of ion, which allows one to establish the molecular mass of peptides. Different mathematical models based on the quantitative structure–property relationship (QSPR) in CZE have been proposed in order to predict the relationship between electrophoretic behavior and structural descriptors (usually from their primary structure, e.g., size or charge) [20]. However, in the case of similar peptides and different buffer systems, semiempirical models are suitable after simplification of the Stokes law given as
where Q is the charge, R is the radius of a spherical object, and η is the Newtonian viscosity of the medium. The first model was proposed by Offord [21], and is expressed as follows:
where Q is the net charge of the peptide, M is its molecular mass, k is an empirical constant, and n is 1 or 2 depending on whether the electrophoretic mobility is expected to be proportional to the volume or surface area of the spherical object investigated [22]. Such an equation can be used for molecular mass calibration of the CZE method similarly to the method established for SEC, where peptide charge data are not necessary (Fig. S1). However, only homologues of PCs are concerned.
The main aim of this work was to create a separation method for studying complexation of cadmium with GSH and PCs with special emphasis on the formation of MMW and HMW complexes. As the stability of the cadmium complexes is a critical factor, CZE was selected as a favorable separation technique for studies of weak interactions between biomolecules [22]. To the best of our knowledge, this work represents the first report on assaying Offord’s model as applied to metal complexes with peptides using CZE ICP MS and showing the influence of metal ions on the exclusion range of the SEC column typically used to indicate the presence of HMW complexes.
Experimental
Chemicals
All chemicals and reagents used were of analytical grade. Ultrapure standards of CdCl2, Trizma, N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (Hepes), and monosodiated and disodiated phosphate were obtained from Fluka (Seelze, Germany). Methanol and sodium hydroxide were purchased from POCh (Gliwice, Poland). Ammonium acetate, β-mercaptoethanol (BMSH), GSH, and proteins for calibration of the SEC column were products of Sigma (Buchs, Switzerland). Standards of PCs of 95 % high-performance liquid chromatography (HPLC) purity were obtained from AnaSpec (Fremont, CA, USA).
Deionized water (18 mΩ cm) prepared with a Mili-Q system (Elix 3; Millipore, Saint-Quentin, France) was used throughout the study.
Sample preparation
Standards of PCs and GSH were prepared by dissolving 1 mg of powder in 1 ml of deionized water (or 5 mM ammonium acetate, pH 7.8 for ESI MS experiments). Dissolved PC samples were divided into small portions (200 μl), purged with an argon stream to prevent oxidation, and eventually frozen for storage. The solutions of PCs were thawed directly before analysis. The stock solution of cadmium ions was prepared weekly by dissolving the appropriate amount of CdCl2 in deionized water. The mixtures of PCs with cadmium ions for SEC and CZE experiments were prepared by dilution of PCs in 20 mM Hepes buffer (pH 7.5) (optionally with 5 mM BMSH) and with addition of cadmium solution to obtain a 1:1 molar ratio.
A weighed sample of 40 mg of freeze-dried plant material (roots of Arabidopsis thaliana L. exposed to cadmium during hydroponic growth), obtained by means of an Alpha 1–2 LD lyophilizator (Christ, Osterode, Germany), was additionally frozen in liquid nitrogen (−196 °C) in order to break cell walls and improve the effectiveness of grinding. Subsequently, 20 mg of pure quartz sand was added, and the sample was ground with a mortar and pestle until a homogenous powder formed. The water-soluble cadmium complexes were extracted from the homogenized sample using 1-ml portions of 20 mM Hepes (pH 7.5), followed by sonication for 30 min. After leaching, the solution was centrifuged twice for 15 min at 15,000 rpm. The supernatant was filtered with a 0.22-μm syringe filter (Sigma-Aldrich, Bellefonte, PA, USA); the first two drops were discarded, leaving only the remaining part of the filtrate to be analyzed by ICP MS and injected into the SEC column or the CZE capillary.
Apparatus
The cadmium complexes were characterized by means of LC MSD 1100 ESI MS and ESI MS/MS 6460 spectrometers (Agilent Technologies, Wilmington, NC, USA) equipped with quadrupole mass analyzers. Calculation of isotope patterns was done with IsoPro 3.0 MS/MS.
CZE experiments were performed with an HP CZE system (Agilent Technologies, Waldbronn, Germany) equipped with an on-column diode-array detector. For all the separations, fused-silica capillaries of 68-cm total length (60-cm effective length; 75-μm inner diameter) from Composite Metal Services (Hallow, UK) were used. The capillary and the sample tray were air-thermostatted at 25 °C. Instrument control and data analysis were done using ChemStation.
SEC was implemented using an HP 1100 gradient HPLC pump (Agilent Technologies, Waldbronn, Germany) with a model 7725 injection valve as the sample delivery system for a sample volume of 50 μl. A model 7500a ICP mass spectrometer (Agilent Technologies, Tokyo, Japan) was used for quantification of the metal content in extracts and for online metal monitoring in the HPLC eluate. The sample or the mobile phase from the SEC column was introduced into the ICP MS system through a Babington nebulizer fitted in a commercial Scott spray chamber.
An ICP mass spectrometer was also coupled to a CZE system (Agilent Technologies, Waldbronn, Germany) through the CZE ICP MS interface based on a microconcentric nebulizer (MCN-100; CETAC, Omaha, NE, USA). The interface was described in detail elsewhere [23, 24]. Briefly, the self-aspirated makeup liquid (2 mM Hepes, pH 7.4 with 10 μg mL-1 germanium added to control the efficiency of the nebulization and stability of the ICP MS performance), grounded by a platinum electrode, was mixed with the separation buffer in a T-connector assembled at the outlet of the CZE capillary. A torch with a smaller inlet (1.5 mm) was used to decrease the influence of plasma backpressure on the electroosmotic flow (EOF) and a platinum shield was installed on the torch to improve sensitivity.
The pH measurements were performed using an Elmetron (Zabrze, Poland) pH meter. A Sonorex ultrasonic bath (model 1210; Bandelin, Walldorf, Germany) was used for leaching and degassing of the solutions.
ESI MS experiments
The mixture of cadmium (5 × 10-4 M) and peptide solutions at a molar ratio of 1:1 was prepared in 5 mM ammonium acetate (pH 7.8) and divided into five portions: first, prior to immediate analysis by ESI MS and next, after incubation for 10, 20, 30, and 60 min at 37 °C. Ammonium acetate was chosen as the buffer because of its good volatility and protonation capacity (in spite of its weak buffering strength at cytosol-like pH).
Optimization of ESI source parameters was done using standard solutions of bioligands and their mixtures with cadmium admitted to the detector by a syringe pump (10 μL min−1). Only the orifice voltage was found to influence the stability of the complexes. The lowest voltage (60 V) was applied to prevent complex decomposition and to ensure efficient transport of the generated ions to the analyzing module. ESI MS spectra were acquired in the ranges 150–1,500 amu (model HP 1100) and 700–2000 amu (model 6460) using a dwell time of 20 ms and an acquisition step of 0.1. An ion spray voltage of 3,500 V was applied for positive ion mode, and 2,500 V was applied for negative ion mode.
Size-exclusion chromatography
A Superdex Peptide HR 10/30 300 mm × 10 mm column (GE Healthcare, Piscataway, NJ, USA) with an exclusion limit of 10 kDa was calibrated by injecting standard solutions of thiopeptides such as GSH (M r 307), PC2 (M r 539), PC3 (M r 771), PC4 (M r 1,003), and apometallothionein II (M r ~ 6,000) and their complexes with cadmium ions prepared in vitro. The column was washed everyday for 30 min with 20 mM Hepes buffer with 5 mM BMSH and 2 mM EDTA (pH 7.4) in order to remove metal ions sorbed on the stationary phase. Sodium chloride (10 mM) was added to Hepes buffer as a noncomplexing salt in order to suppress the ion interactions during the chromatographic process. The isotopes (112Cd and 111Cd) were monitored to observe changes in the isotopic profiles and control their ratio in case of any interferences.
The chromatographic recovery for cadmium complexes was calculated as the ratio of the cadmium concentration in the SEC eluate (about 5 mL) against the cadmium concentration in the injected sample regarding the appropriate dilution.
CZE experiments
Fused silica was activated by the rinsing the CZE capillary with 1 M NaOH and Milli-Q water for 15 min. The electrolyte vials were replenished after each analysis and the capillary was rinsed with Milli-Q water, 0.1 M NaOH, Milli-Q water, and electrolyte solution (each for 2 min). A solution of 20 mM Hepes (pH 7.5) was used as a background electrolyte (BGE). The level and stability of the current (9.2 ± 0.2 μA at 25 kV) guaranteed effective temperature control (25 °C) by an air-thermostatic system. Samples were introduced using the hydrodynamic method at 80 mbar s, which corresponds to a volume of about 40 nl.
The recovery of cadmium from the CZE capillary was calculated from the amount of cadmium not adsorbed on the capillary wall: (y - x/y) × 100 %, where x is the peak area obtained by rinsing the CZE capillary with BGE after the electrophoretic process and y is the total peak area obtained by rinsing the CZE capillary directly after injection of the sample. The flow during flushing of the capillary was about 1 μl min-1, which corresponds to the level of the established EOF.
Results and discussion
As there are no standards for cadmium complexes with PCs, the structure of the complexes created in vitro needed to be examined by ESI MS. Molecular masses for the complexes established on the basis of the most intense signals were necessary for molecular mass calibration of the SEC column and the CZE method.
Establishment of the stoichiometry of LMW complexes by ESI MS
The investigated mixtures of bioligands with CdCl2 · in a 1:1 molar ratio were injected directly into the ESI source, which is known to generate multicharged ions especially in the case of proteins and peptides. This phenomenon was also observed in the case of cadmium complexes of thiopeptides for both negatively charged and positively charged ions (Table 1). However, the signals obtained for the investigated peptides in negative ion mode at pH 7.8 were usually ten times higher because of favored deprotonation of the carboxylic group.
As the complexity of the mass spectra was high, they were analyzed in four steps, by establishing:
-
1.
The charge of the ion
-
2.
The number of cadmium ions attached to thiopeptide by checking the agreement between the theoretical and the experimental isotopic profile
-
3.
The number of hydrogen molecules lost during cadmium binding (one H2 per Cd ion [18]) or oxidation of peptide
-
4.
The agreement between the signals observed in positive and negative ion modes
Signals corresponding to 1:1 stoichiometry were dominant (relative abundance from 40 to 100 % in the selected ranges shown in Fig. 1), but PC2 and PC4 were found to bind two cadmium ions as well, and signals corresponding to Cd2L were observed in positive and negative ion modes to some extent (relative intensity of up to 20 % in the mass spectra). However, CdL complexes appeared to be stabler and were observed even after 60 min of incubation. Signals corresponding to Cd2L and Cd1L2 were usually not observed after 40 min of incubation; instead, the intensity of signals corresponding to oxidized forms of GSH and PCs increased. Although neither the formation of PC complexes with cadmium nor oxidation of peptides in the ESI source can be excluded, the changes in the intensity of signals with the time for which the samples were incubated indicate that formation of disulfide bridges occurs mainly in solution. Additionally, signals corresponding to oxidized peptides and HMW complexes were not observed in the mass spectra for the peptide mixture with cadmium ions injected directly after preparation. This proves the low significance of the oxidation process in the ESI source. It can be concluded that the presence of metal ions enhances the oxidation of thiopeptides in solution, which was already reported by other authors [8, 12, 13].

Mass spectra obtained in negative ion mode for mixture of Cd with a glutathione (GSH), b PC2 (PC is phytochelatin), c PC3, and d PC4 (100 μg ml-1) in 1:1 molar ratio prepared in 10 mM ammonium acetate, pH 7.5 after 20 min of incubation. The comparison of experimental and theoretical isotopic patterns for selected ions is presented in the insets of the mass spectra (c, d)
With the progress of oxidation of PCs, new HMW complexes were observed, especially in the case of PC2. Although the interpretation of many signals in the mass spectra was hampered by the complexity of the isotopic profile in the case of multicharged species, the agreement between the experimental and the theoretical isotopic profile was above 70 % for interpreted signals. PC2 was found to create Cd3(PC2)2 (M exp 1,410.0), Cd4(PC2)2 (M exp 1,522.0), Cd4(PC2)3 (M exp 2,061.2), andCd5(PC2)3 (M exp 2,175), which can be produced with the disulfide bridges between two or even three molecules of the peptide. Although HMW complexes (M r > 2,000) were not identified in the case of PC3 and PC4, their ability to create multicharged species cannot be excluded. Moreover, the mass spectra were rich in signals at the level of noise (relative abundance of about 1–2 %), containing dense clusters with interspaces such as 0.2 or 0.3 u. Additionally, several signals observed in the mass spectra of PC3 corresponding to oxidized multicharged species of L3 and L5 indicate that HMW complexes should be created. It is possible that their detection was hindered by the sensitivity of the mass spectrometer or the narrow mass range (up to 2,000 u) of the quadrupole mass analyzer. As quadrupole mass analyzers offer low accuracy for establishing the molecular mass of compounds (from 100 ppm to almost 900 ppm), the identity of the complexes needed to be verified by MS/MS experiments. The fragmentation was done for the most intense negatively charged ions using a collision energy from 10 to 40 eV, which was high enough to obtain signals complementary to those for ligands released from the complexes. For instance, signals at m/z 289 and 596 were obtained by fragmentation (at 15 eV) from signals at m/z 418 and 725 by loss of a Glu residue (−129 Da). The main product ion was observed at m/z 306. Signal, typical for the GSSG conjugate, such as that at m/z 467 obtained from m/z 725 through the loss of two Glu residues (−258 Da) was not found [25]. Other specific product ions of collision-induced dissociation expected at m/z 338 or 450 corresponding to the RSSH moiety and its complex with cadmium ion and at m/z 272 produced by loss of an H2S2 molecule (−66 Da) [25] were not observed either. However, signals at m/z 272 and 338 were observed for a parent ion at m/z 611 corresponding to [GSSG - H]-. The most interesting MS/MS results were obtained for the m/z 1,029.5 parent ion, corresponding to Cd4(PC2)3. The following signals were obtained for 20 eV (see Fig. S2): at m/z 1,945.2 produced through released (114 Da) cadmium ion and H2, at m/z 1,677.5 produced by further loss of the Glu-Cys fragment and two molecules of H2O; at m/z 1,798.2 obtained by loss of a Glu residue and H2O; and the most intense at m/z 538 corresponding to PC2. Other signals at m/z 1,400.3 and 1,169.8 could correspond to Cd2(PC2)1-GlyGlu and (Cd(PC2)2 - H2O), respectively. However, their identity should be verified by other MS/MS experiments using an ion trap. It was impossible to provide unambiguous proof of the presence of disulfide bridges by collision-induced-dissociation experiments, even for higher collision energies (30, 40 eV). Specific release of cadmium ion with a hydrogen molecule from Cd4(PC2)3 could indicate two phenomena: first, disulfide bridges are created through the metal ion released from the complex; second, ionic interactions between metal ions and polar groups such as carboxylic or amine groups might be decisive for the formation of MMW (0.8–2.0 kDa) and HMW cadmium complexes.
Characterization of the SEC method
The first method chosen for preliminary characterization of the metal complexes was SEC because of gentle steric interactions. Nevertheless, the presence of anionic residues can cause other unspecific but stronger interactions, leading to complex degradation. Firstly, 20 mM ammonium acetate (as ESI MS friendly) was considered, but the recovery of cadmium was below 50 % and there were only four peaks. The best total chromatographic recovery established for cadmium was 60 %, when 20 mM tris(hydroxymethyl)aminomethane–HCl and Hepes–NaOH were used. It was necessary to add NaCl (10 mM) to the mobile phase to obtain recovery up to 80 %. Another SEC column with a diol stationary phase was also examined (Zorbax; Agilent Technologies). In the case of the silica-modified stationary phase, 100 mM NaCl was necessary to obtain acceptable cadmium recovery. Such a high concentration of salt in the mobile phase was responsible for noticeable deterioration of the torch inlet tube after only 4 h of work. As a result, a column with a polydextran stationary phase was selected as more suitable.
The recovery of cadmium from the stationary phase was also found to depend significantly on the concentration of the complexes (80–90 % down to 50 μg ml-1 thiopeptides, and 30 % for 100 ng ml-1). This means that saturation of the stationary phase with cadmium ions improves metal recovery and the sensitivity of SEC ICP MS. However, the method is not reliable in the case of plant extracts of different origin, blank samples, and especially for the control group of cultivated plants not exposed to metal stress. The method could be applied provided a careful and rigorous washing protocol was employed.
The same mixtures of thiopeptides with cadmium were injected into the SEC column after different incubation times. The recovery of cadmium decreased with increasing time for which the sample was incubated in ambient conditions and new peaks 1–4 in Fig. 2 were observed. However, these differences were observed after at least 30 min of incubation.

Size-exclusion chromatography (SEC) inductively coupled plasma mass spectrometry (ICP MS) chromatograms obtained for mixtures of glutathione and phytochelatin complexes (concentration 50 μg ml-1 each) after 5 and 60 min of incubation. The calibration curves for the Superdex Peptide HR 10/30 column are shown below the chromatograms: squares apometallothionein and free thiopeptides, crosses cadmium-bound peptides. Peaks numbered 1–4 correspond to data in Table 2
Peaks corresponding to LMW cadmium complexes were identified as Cd1L1 (Fig. 2) by internal standard addition of a single thiopeptide. Their intensity decreased with time as observed during ESI MS experiments (Fig. 2). The addition of the reducing agent (BMSH) to the sample was responsible for higher recovery of cadmium (up to 90 %) and an increase of height of the chromatographic signals identified as Cd1L1 complexes. Such behavior can be explained by retrieval of reactive thiol groups, which causing strong binding of cadmium ions. Still, the height of the peaks observed in the MMW and HMW fractions (peaks 1–4 in Fig. 2) decreased slightly (not shown).
SEC provides the possibility of establishing the molecular mass of separated compounds. Compounds for column calibration are typically selected in order to define all the exclusion range (appropriate polymers and proteins). However, in the case of cadmium complexes, ionic interactions should be taken into consideration.
A Superdex Peptide HR 10/30 column was calibrated with two mixtures: apometallothionein and free PCs (with UV detection, 214 nm) and using a solution of a mixture of cadmium with the thiopeptides investigated (with ICP MS for detection, 111Cd). The parameters for the SEC column obtained with free peptides and apometallothionein were in agreement with the parameters provided by the manufacturer. The exclusion limit was 10 kDa and the optimal separation range was 0.3–7.0 kDa. However, calibration conducted with cadmium complexes showed a change in method selectivity (significant change of the slope of the calibration graph in Fig. 2) and a change in the optimal separation range to a narrower range (0.4–2.0 kDa). This means that for calibration of SEC columns, compounds possibly similar to the ones investigated should be used. Although the repeatability of the retention times was satisfactory (relative standard deviation below 2 %), the accuracy of the established molecular mass is insufficient to propose the stoichiometry of the complexes (Table 2). Nevertheless, the molecular masses obtained were not as high as expected, leading to the conclusion that peaks 2–4 in Fig. 2 belong to MMW complexes instead of HMW complexes. Moreover, the molecular masses established for peaks 2 and 3 were in good agreement with the molecular masses of two complexes found by ESI MS: Cd3(PC2)2—M exp 1,410.2; Cd4(PC2)2—M exp 1,522.0. However, other adducts created by different thiopeptides (e.g., Cd3GSH-PC3, Cd3GSH2-PC2) cannot be excluded.
The SEC column was coupled to an ESI MS detection system, and a solution of 10 mM ammonium acetate, pH 7.4 (0.5 ml min-1) was used as the mobile phase. Methanol was added as a makeup flow (0.1 ml min-1) following the protocol previously established for GSH and amino acid complexes with metal ions [12]. Since ESI MS offers lower sensitivity than ICP MS, especially in scan mode (in the range 700–2,000 m/z), selected ion monitoring was performed by registering the ions described in Table 1. Additionally, ions corresponding to different thiopeptide conjugates were included (e.g., PC2–4-SG, PC2–4-PC2–3). In spite of a shift in retention times and loss of resolution in comparison with a Hepes mobile phase, it was possible to confirm the identity of the complexes eluted as the last four peaks. Moreover, it was found that Cd1(GSH)2 was coeluted with Cd1(PC2)1. However, it was impossible to establish the stoichiometry of the complexes eluted as peaks 1–4 (Fig. 2), probably owing to the low efficiency of the ESI process. Interestingly, it was possible to reconstruct chromatograms corresponding to GSH and PC conjugates (i.e., at m/z 843, 1,073 and 1,075, 1,305 and 1,307, 1,535 and 1,537, and 1,769, corresponding to PC2-SG, PC3-SG–PC2PC2, PC4-SG–PC3-PC2, and PC4-PC3, respectively), and their elution times were inversely proportional to their molecular mass.
The nature of peptide’s behavior during CZE experiments
CZE UV experiments were performed for a standard mixture of peptides to characterize the nature of their behavior during electrophoretic separation. Ammonium acetate as a BGE was responsible for dispersion of the peaks, long analysis time (25 min), and in consequence low resolution and signal intensity related to low cadmium recovery (62–71 %). On the other hand, tris(hydroxymethyl)aminomethane allowed the analysis time to be shortened to 3 min, but the resolution was substantially lower, with only two peaks being observed. Hepes and phosphate offered the best shape of the electrophoretic peaks and the highest recovery, 92–97 %. Moreover, Hepes has been reported not to influence the conformation of proteins and peptides and not to interact with metal ions [26]. However, its zwitterionic character can be responsible for interactions with negatively charged silanol groups and high recovery of cadmium even if a lower concentration of peptides is injected (87 % for 100 ng ml-1). The “covering effect” of Hepes toward silanol groups prevents sorption of both metal ions and peptides, which can lead to subtle separation conditions with a diminished influence on metal–peptide equilibrium.
The charge of peptides in cytosol-like BGE conditions was negative, because signals were observed after migration of the peak for methanol added to the sample as an EOF marker (Fig. 3a). This effect is in agreement with the report of Dorcak and Krezel [18]. The order of migration from the smallest to the biggest peptide suggests that the negative charge increases with the molecular mass and the number of polar acidic groups. The influence of the charge and molecular size of the peptide on its effective mobility was checked for different semiempirical models. The net charge of each peptide was calculated using the Henderson–Hasselbach equation [27] and was −1.0, -2.1, -3.1, and −4.2 for GSH, PC2, PC3, and PC4, respectively. As expected, the agreement between the results obtained and the semiempirical model suggested by Offord was better in the case of interactions proportional to the surface area (r 2 = 0.999) than to the volume (r 2 = 0.995).

Electropherograms obtained by capillary zone electrophoresis (CZE) with UV detection for mixtures of phytochelatins and glutathione (50 ppm each) in 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid, pH 7.4 without (a) or with (b) cadmium added to the sample. The identity of the compounds was established by comparison of the migration time for cadmium mixtures with single peptide and internal standard addition. Peaks numbered 1–3 correspond to data in Table 3. BMSH β-mercaptoethanol
The equation obtained for free peptides using calculated net charges was
As it can be impossible to establish the charge for unknown species, the coefficient k can include changes in the charge in the case of similar compounds, and Q was defined as −1 for all peptides. Although the agreement between the results and Offord’s model did not change, the molecular mass established for peptides with different chemical properties would be inaccurate.
After addition of cadmium ions to a mixture of the investigated peptides, the main revelation was the prolongation of the migration time of the compounds. Binding of Cd2+ was expected to compensate the negative charge, leading to migration of complexes before the EOF marker, but the complexes created were still negatively charged (Fig. 3b). However, slower migration in comparison with free peptides indicates that the density of negative charge was higher or the molecule became smaller. Binding of cadmium ion could be responsible for creating a tight coil of peptide around metal without significant changes in the charge on the surface and leading to slower migration, which could explain the nature of the results obtained. The addition of BMSH to the sample improved the repeatability of the results and the precision of the migration times became satisfactory (relative standard deviation 1.5–3.6 %). The peak of the Cd–BMSH complex was observed about 2 min after that of methanol and it was used as a pseudo marker for changes in the EOF when ICP MS was used for detection.
Hence, the CZE UV electropherogram contains about 14 peaks and the selectivity of UV detection does not allow us to distinguish cadmium complexes from nonbound oxidized thiopeptides, and therefore a more specific detection method such as ICP MS was required. As can be seen in Fig. 4, spectra A, the number of peaks obtained by CZE ICP MS was lower than during UV detection. Again, the agreement for migration of the complexes with Offord’s model was checked for pseudo-electrophoretic mobility established using Cd–BMSH as a marker for changes in the EOF:

Electropherograms obtained by CZE ICP MS for mixtures of phytochelatins and glutathione (50 ppm each) with Cd2+ in 1:1 molar ratio. Mixtures of GSH, PC2, PC3, and PC4 (A), GSH and PC2 (B), PC2 and PC3 (C), and PC3 and PC4 (D) with Cd2+ after 30 min of incubation. Peaks numbered 1–3 correspond to data in Table 3
Although the selectivity and mass range for CZE ICP MS proved to be more accurate in comparison with SEC ICP MS, the resolution was still too low (standard deviation ±50–200 Da) to propose the stoichiometry of the complexes created. Taking into account the fact that the mass difference between the thiopeptides is 232 Da and two cadmium atoms give 228 Da, the standard deviation for the established molecular mass should be ±4 Da, which is impossible to obtain by any separation technique, especially one based on extrapolation (as in the case of CZE). Another problem which appeared was the spreading of the last two peaks (peaks 2 and 3 in Fig. 4, spectra A), which could be caused by typical electrophoretic peak dispersion increasing with the migration time. It could also be caused by co-migration of many created species. Mixtures of cadmium ions with single thiopeptides were found to create at least MMW complexes (not shown), but the mixture of two PCs created even more species after 30 min of incubation. Diversity of the complexes was observed in the case of all the thiopeptides (Fig. 4, spectra B–D). It could originate from the created conjugates of thiopeptides. However, in the presence of metal ions, the creation of agglomerates through ionic interaction with metal ions should be considered as well.
Any created species that migrated after the Cd1L1 complex could indicate that the m ef of the created cadmium complexes is still proportional to the surface area of created species and their molecular mass can be estimated by the CZE method. Equation 4 was used to calculate the molecular masses for the peaks indicated in Figs. 3 and 4 (Table 3).
The role of oxidation of PCs was also examined. BMSH prolonged the creation of agglomerates. For example, the incubation needed to be conducted for at least 30 min when 10 mM BMSH was present in the solution. When BMSH was not added to the sample, peaks 2 and 3 (Fig. 4) were observed after 10 min of incubation.
Although the creation of multicharged species cannot be excluded in the case of multicomponent mixtures, the CZE ICP MS method shows good selectivity to examine polymorphism of Cd–thiopeptide complexes. One of the most important limitations of this technique is the lack of the possibility to perform unambiguous identification of separated species, which could be provided by ESI MS. However, both SEC ICP MS and CZE ICP MS can be useful to establish an appropriate protocol for sample preparation.
Characteristic of cytosol extract
The extract of Arabidopsis thaliana L. exposed to cadmium stress was prepared for both SEC ICP MS analysis and CZE ICP MS analysis. The extraction protocol with 20 mM Hepes buffer enhanced with sonication was selected in order to obtain good recovery of cadmium species (44 % of the total amount of cadmium was established for roots by ICP MS) and to increase the probability of high species diversity with different stability. The recovery of cadmium from the stationary phase was checked and was established as 87 % and 95 % for SEC and CZE, respectively. However, sensitivity in the case of SEC was 100 times higher owing to the much bigger (about 1,000 times) sample volume used for analysis.
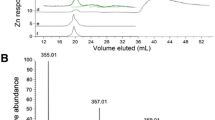
The limitations of SEC in comparison with CZE were also observed on using the plant extracts. Firstly, the electropherogram obtained for the control sample of a plant not exposed to cadmium did not contain any peaks. The chromatogram obtained by SEC ICP MS contained peaks of cadmium traces extracted by sample components from the stationary phase. Secondly, except for complexes of single thiopeptides with Cd2+, four additional peaks were found by CZE ICP MS, whereas only two of them were observed using SEC ICP MS. Two fractions were collected from the SEC column (Fig. 5, top) and after lyophilization were dissolved in BGE, centrifuged, filtered, and analyzed by CZE ICP MS. For the first chromatographic fraction (more than 2.1 kDa by SEC), four different electrophoretic peaks were observed by CZE (Fig. 5, bottom) but the major ones were peaks 2, 3, and 4, corresponding to complexes with molecular masses of 2.6, 3.6, and 6.0 kDa, respectively, as established by the CZE ICP MS method. For the second SEC fraction (1.6 kDa by SEC), the electrophoretic peak indicated as 1 with a molecular mass of 1.6 kDa established by CZE ICP MS was dominant. Such agreement between size-exclusion fractionation and electrophoretic migration confirms the potential of CZE coupled with ICP MS as a suitable technique for characterization of MMW and HMW complexes created by Cd–PC complexes. However, the quality of the results strongly depends on the sample preparation protocol, which affects both the level of oxidation of PCs and the stoichiometry of their complexes with cadmium ions. It is impossible to determine whether the speciation measured in the extract is representative of specific equilibrium in the plant’s cytosol. Nevertheless, the disturbance of the metal–peptide equilibrium caused by the separation process should be limited and controlled at least by recovery of metal from the stationary phase.

SEC ICP MS chromatogram (top; chromatogram in black obtained for a plant not exposed to cadmium) and CZE ICP MS electropherogram (bottom) obtained for an extract of Arabidopsis thaliana exposed to Cd2+. Peaks I and II correspond to fractions collected from the SEC column and analyzed by CZE ICP MS. Molecular masses established by the CZE method for unknown cadmium complexes are indicated in the electropherogram: 1 1.6 kDa, 2 2.6 kDa, 3 3.6 kDa, and 4 6.0 kDa
Conclusions
Both the SEC ICP MS method and the CZE ICP MS confirmed the ability of thiopeptides to create MMW and LMW complexes with cadmium ions and proved the presence of disulfide bridges to be one of the factors connected to their formation. Although the selectivity of the CZE method is considerably better in comparison with that of the SEC method, the resolution is insufficient to establish the stoichiometry of the complexes. Moreover, the CZE method is more suitable to examine HMW complexes because its exclusion range is less narrow in comparison with the SEC method. However, CZE (especially coupled with ICP MS) is strongly sensitive to matrix effects. Hence, both SEC ICP MS and CZE ICP MS should be treated as complementary for studies of polymorphism of Cd–thiopeptide complexes.
References
Wood BA, Feldmann J (2008) Quantification of phytochelatins and their metal(loid) complexes: critical assessment of current analytical methodology. Anal Bioanal Chem 390:1753–1764
Zenk MH (1996) Heavy metal detoxification in higher plants–a review. Gene 179:21–30
Cobbett C, Goldsbrough P (2002) Phytochelatins and metallothioneins: roles in heavy metal detoxification and homeostasis. Annu Rev Plant Biol 53:159–182
Wesenberg D, Krauss G-J, Schaumloffel D (2011) Metallo-thiolomics: investigation of thiol peptide regulated metal homeostasis in plants and fungi by liquid chromatography-mass spectrometry. Int J Mass Spectrom 307:46–54
Vatamaniuk OK, Mari S, Lu YP, Rea PA (2000) Mechanism of heavy metal ion activation of phytochelatin (PC) synthase: blocked thiols are sufficient for PC synthase-catalyzed transpeptidation of glutathione and related thiol peptides. J Biol Chem 275:31451–31451
Gusmão R, Ariño C, Díaz-Cruz JM, Esteban M (2010) Electrochemical survey of the chain length influence in phytochelatins competitive binding by cadmium. Anal Biochem 406:61–69
Mutoh N, Hayashi Y (1988) Isolation of mutants of Schizosaccharomyces pombe unable to synthesize cadystin, small cadmium-binding peptides. Biochem Biophys Res Commun 151:32–39
Vacchina V, Połeć K, Szpunar J (1999) Speciation of cadmium in plant tissues by size-exclusion chromatography with ICP-MS detection. J Anal At Spectrom 14:1557–1566
Połeć-Pawlak K, Ruzik R, Abramski K, Ciurzyńska M, Gawrońska H (2005) Cadmium speciation in Arabidopsis thaliana as a strategy tostudy metal accumulation system in plants. Anal Chim Acta 540:61–70
Bräutigam A, Wesenberg D, Preud'homme H, Schaumlöffel D (2010) Rapid and simple UPLC-MS/MS method for precise phytochelatin quantification in alga extracts. Anal Bioanal Chem 398:877–883
Chen L, Guo Y, Yang L, Wang Q (2007) SEC-ICP-MS and ESI-MS/MS for analyzing in vitro and in vivo Cd-phytochelatin complexes in a Cd-hyperaccumulator Brassica chinensis. J Anal At Spectrom 22:1403–1408
Połeć-Pawlak K, Ruzik R, Lipiec E (2007) Investigation of Cd(II), Pb(II) and Cu(I) complexation by glutathione and its component amino acids by ESI-MS and size exclusion chromatography coupled to ICP-MS and ESI-MS. Talanta 72:1564–1572
Yen T-Y, Villa JA, DeWitt JG (1999) Analysis of phytochelatin–cadmium complexes from plant tissue culture using nano-electrospray ionization tandem mass spectrometry and capillary liquid chromatography/electrospray ionization tandem mass spectrometry. J Mass Spectrom 34:930–941
Sadi BBM, Vonderheide AP, Gong J-M, Schroeder JI, Shann JR, Caruso JA (2008) An HPLC-ICP-MS technique for determination of cadmium-phytochelatins in genetically modified Arabidopsis thaliana. J Chromatogr B 861:123–129
Gusmão R, Cavanillas S, Ariño C, Díaz-Cruz JM, Esteban M (2010) Circular dichroism and voltammetry, assisted by multivariate curve resolution, and mass spectrometry of the competitive metal binding by phytochelatin PC(5). Anal Chem 82:9006–9013
Gusmão R, Ariño C, Díaz-Cruz JM, Esteban M (2010) Cadmium binding in mixtures of phytochelatins and their fragments: a voltammetric study assisted by multivariate curve resolution and mass spectrometry. Analyst 135:86–95
Leverrier P, Montigny C, Garrigos M, Champeil P (2007) Metal binding to ligands: cadmium complexes with glutathione revisited. Anal Biochem 371:215–228
Dorcak V, Krezel A (2003) Correlation of acid–base chemistry of phytochelatin PC2 with its coordination properties towards the toxic metal ion Cd(II). Dalton Trans 11:2253–2259
Chekmeneva E, Gusmão R, Díaz-Cruz JM, Ariño C, Esteban M (2011) From cysteine to longer chain thiols: thermodynamic analysis of cadmium binding by phytochelatins and their fragments. Metallomics 3:838–846
Mittermayr S, Olajos M, Chovan T, Guttman A (2008) Mobility modeling of peptides in capillary electrophoresis. Trends Anal Chem 27:407–417
Offord RE (1966) Electrophoretic mobilities of peptides on paper and their use in the determination of amide groups. Nature 211:591–593
Girardot M, Gareil P, Varenne A (2010) Interaction study of a lysozyme binding aptamer with mono and divalent cations by ACE. Electrophoresis 31:546–555
Prange A, Schaumlöffel D (1999) Determination of element species at trace levels using capillary electrophoresis-inductively coupled plasma sector field mass spectrometry. J Anal At Spectrom 14:1329–1332
Połeć-Pawlak K, Abramski JK, Ferenc J, Foteeva LS, Timerbaev AR, Keppler BK, Jarosz M (2008) Application of capillary electrophoresis-inductively coupled plasma mass spectrometry to comparative studying of the reactivity of antitumor ruthenium(III) complex differing in the nature of counter-ion toward human serum proteins. J Chromatogr A 1192:323–326
Thakur S, Padanabhan B (2008) Fragmentation of peptide disulfides under conditions of negative ion mass spectrometry: studies of oxidized glutathione and contryphan. Am J Soc Mass Spectrom 19:358–366
Kragh-Hansen U, Vorum H (1993) Quantitative analyses of the interaction between calcium ions and human serum albumin. Clin Chem 39:202–208
Skoog B, Wichman A (1986) Calculation of the isoelectric points of polypeptides from the amino acid composition. Trends Anal Chem 5:82–83
Acknowledgments
K.P. is grateful to Warsaw University of Technology for financial support of the project realized within COST action CM0902. A.M. is grateful for the financial support of the European Union in the framework of the European Social Fund through the Warsaw University of Technology Development Programme, realized by the Center for Advanced Studies and Mazovia PhD Scholarship, funded by the Office of the Marshal of the Mazowiecki Voivodeship from Warsaw.
Author information
Authors and Affiliations
Corresponding author
Additional information
ABC Highlights: authored by Rising Stars and Top Experts
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 3771 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Miszczak, A., Rosłon, M., Zbroja, G. et al. SEC ICP MS and CZE ICP MS investigation of medium and high molecular weight complexes formed by cadmium ions with phytochelatins. Anal Bioanal Chem 405, 4667–4678 (2013). https://doi.org/10.1007/s00216-013-6868-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-6868-3