
Abstract
Aims/hypothesis
Cholecystokinin (CCK) and leptin are important hormones with effects on energy balance. The present study assessed the biological effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3, smaller isoforms of CCK and leptin, respectively.
Methods
The actions and overall therapeutic use of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3, alone and in combination, were evaluated in normal and high-fat-fed mice.
Results
(pGlu-Gln)-CCK-8 had prominent (p < 0.01 to p < 0.001), acute feeding-suppressive effects, which were significantly augmented (p < 0.05 to p < 0.01) by [d-Leu-4]-OB3. In agreement, the acute dose-dependent glucose-lowering and insulinotropic actions of (pGlu-Gln)-CCK-8 were significantly enhanced by concurrent administration of [d-Leu-4]-OB3. Twice daily injection of (pGlu-Gln)-CCK-8 alone and in combination with [d-Leu-4]-OB3 in high-fat-fed mice for 18 days decreased body weight (p < 0.05 to p < 0.001), energy intake (p < 0.01), circulating triacylglycerol (p < 0.01), non-fasting glucose (p < 0.05 to p < 0.001) and triacylglycerol deposition in liver and adipose tissue (p < 0.001). All treatment regimens improved glucose tolerance (p < 0.05 to p < 0.001) and insulin sensitivity (p < 0.001). Combined treatment with (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 resulted in significantly lowered plasma insulin levels, normalisation of circulating LDL-cholesterol and decreased triacylglycerol deposition in muscle. These effects were superior to either treatment regimen alone. There were no changes in overall locomotor activity or respiratory exchange ratio, but treatment with (pGlu-Gln)-CCK-8 significantly reduced (p < 0.001) energy expenditure.
Conclusions/interpretation
These studies highlight the potential of (pGlu-Gln)-CCK-8 alone and in combination with [d-Leu-4]-OB3 in the treatment of obesity and diabetes.
Similar content being viewed by others


Introduction
Peripheral signals that control food intake and energy expenditure encompass a number of factors including adipokines, insulin and a variety of gastrointestinal derived hormones [1]. In this context, cholecystokinin (CCK) and leptin are two important peptides with pertinent effects on feeding and body weight control [2]. CCK is secreted from intestinal I cells in response to fatty acids and proteins in the intestinal lumen [3]. It is widely acknowledged that the major physiological role of CCK is in the short-term stimulation of satiety [4]. Specifically, the actions of CCK on food intake are mediated peripherally by CCK1 receptors on vagal afferent neurons [4]. Indeed, peripherally released CCK cannot penetrate the blood–brain barrier [5]. There are a number of molecular isoforms of CCK ranging from 58 to four amino acids in length, but CCK-8 is the smallest form that retains the full range of biological actions [6]. Thus, we have developed (pGlu-Gln)-CCK-8, an enzymatically stable CCK-8 analogue, and shown that sub-chronic twice daily administration of this peptide can reverse many of the established metabolic abnormalities associated with obesity/diabetes [7].
Although CCK regulates energy balance in the short term, its actions on longer-term energy control are less discernible. Thus, in the long term ‘adiposity signals’ play a major role in determining energy stores within the body, the most important of which is the peptide leptin [8]. Leptin, secreted into the circulation from white adipose tissue and gastric endocrine cells, alters food intake and energy expenditure to help continually maintain an appropriate body weight and level of adiposity [9]. As such, administration of exogenous leptin results in sustained reduced food intake and loss of body weight [10]. Similar to CCK, there is evidence to suggest that the entire leptin molecule is not required for stimulating its biological actions [11, 12]. Thus, a synthetic peptide corresponding to amino acid residues 116–122 of mouse leptin, with an additional substitution of the Leu residue at position 4 with its d-isomer, namely [d-Leu-4]-OB3, retains all significant biological actions of full length leptin [12, 13].
Leptin mediates energy balance by acting through two distinct receptors encoded by the Ob-R gene (also known as LEPR), a short-form Ob-Ra and a long-form Ob-Rb receptor [13]. In addition, there are a number of other isoforms of the leptin receptor that may have physiological significance [14]. The metabolic effects of leptin are generally attributed to activation of receptors in the hypothalamus [15]. However, reports indicate that leptin receptor mRNA is present on vagal afferent neurons [16]. Interestingly, the long form of the leptin receptor has been found to be co-expressed with the CCK1 receptor in a subset of vagal afferents [16]. Thus, CCK- and leptin-derived peptides could have additive therapeutic promise for obesity/diabetes. In this regard, CCK has been shown to facilitate the effect of leptin on decreasing body weight through synergistic effects on reducing food consumption and activation of metabolic pathways related to energy balance [17, 18]. Furthermore, CCK-8 increases the permeability of the blood–brain barrier to leptin [19] and may also induce leptin release from adipose tissue [20]. In harmony with this, leptin has been shown to stimulate CCK secretion [21]. Thus, leptin and CCK may potentiate their own effects by cross-stimulating their secretion. Furthermore, leptin has recently been shown to play a role in modulating the sensitivity of vagal afferents to CCK [22].
Therefore, in the present study we evaluated the relative efficacy and combined therapeutic utility of two promising new compounds for the treatment of obesity/diabetes. The acute dose-dependent individual and combined effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on energy intake, glycaemic control and insulin secretion were assessed in normal mice. In addition, we administered twice daily injections of (pGlu-Gln)-CCK-8 or [d-Leu-4]-OB3, alone and in combination, in high-fat-fed mice to examine the metabolic effects of chronic treatment on body weight, food intake, energy expenditure, non-fasting glucose and insulin, i.p. glucose tolerance, insulin sensitivity, and both tissue and circulating blood lipids.
Methods
Peptides synthesis
(pGlu-Gln)-CCK-8 was obtained from American Peptide Company (Sunnyvale, CA, USA). [d-Leu-4]-OB3 was purchased from GL Biochem (Shanghai, China). Peptides were characterised using matrix-assisted laser desorption ionisation time-of-flight (MALDI-TOF) mass spectrometry [23].
Acute effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 in normal mice
Male Swiss National Institutes of Health (NIH) mice (n = 8) had free access to drinking water and a standard rodent maintenance diet (10% fat, 30% protein and 60% carbohydrate; per cent of total energy of 12.99 kJ/g; Trouw Nutrition, Northwich, UK). In the first series of experiments, 18 h fasted 12- to 15-week-old mice received an i.p. injection of saline alone (0.9% [wt/vol.] NaCl) or in combination with (pGlu-Gln)-CCK-8 (0.2–25 nmol/kg body weight), [d-Leu-4]-OB3 (8.33 mg/kg body weight), or a combination of both peptides. Food intake was measured at 30 min intervals at the times indicated on the figures. In a second series of experiments, similar groups of 4 h fasted mice received an i.p. injection of glucose alone (18 mmol/kg body weight) or in combination with (pGlu-Gln)-CCK-8 (1–25 nmol/kg body weight), [d-Leu-4]-OB3 (8.33 mg/kg body weight), or a combination of both peptides. Plasma glucose and insulin were assessed at the times indicated on the figures.
Longer-term effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 in high-fat-fed mice
Over an 18 day period, male Swiss NIH mice (n = 8) maintained on a high-fat diet (45% fat, 35% carbohydrate and 20% protein; per cent of total energy of 26.15 kJ/g; Special Diet Services, Witham, UK) from 6 weeks of age for 120 days received twice daily i.p. injections (09:00 and 17:00 hours) of saline vehicle (0.9% [wt/vol.] NaCl), (pGlu-Gln)-CCK-8 (25 nmol/kg body weight), [d-Leu-4]-OB3 (8.33 mg/kg body weight) or a combination of both peptides. Food intake and body weight were recorded (10:00 hours) every 3–4 days. An i.p. glucose tolerance test (18 mmol/kg body weight) and an insulin sensitivity test (20 U/kg body weight) were performed at the end of the treatment period. All acute tests commenced at 10:00 hours. Liver, gastrocnemius muscle and subcutaneous adipose tissues were excised at the end of the treatment period without use of anaesthesia and processed for measurement of triacylglycerol content as described previously [24]. Blood lipid profile was also assessed on day 18 using the Friedewald equation to calculate the concentration of LDL-cholesterol [25]. In addition, pancreatic tissue was excised and weighed, and insulin content was determined as described previously [24].
In a separate series, mice were placed in Complete Laboratory Animal Monitoring System (CLAMS) metabolic chambers (Columbus Instruments, Columbus, OH, USA) following the normal 09:00 hours daily injection on day 18. Consumption of O2 and production of CO2 were measured for 30 s at 15 min intervals for a total of 22 h. Respiratory exchange ratio (RER) was calculated by dividing \( \mathop{V}\limits^{.}\mathrm{C}{{\mathrm{O}}_2} \) by \( \mathop{V}\limits^{.}{{\mathrm{O}}_2} \). Energy expenditure was calculated using RER with the following equation \( \left( {3.815+1.232\times \mathrm{RER}} \right)\times \mathop{V}\limits^{.}{{\mathrm{O}}_2} \). Ambulatory locomotor activity of each mouse was measured simultaneously using optical beams (Opto M3, Columbus Instruments). Consecutive photo-beam breaks were scored as an ambulatory movement. Activity counts in X and Z axes were recorded every minute for 22 h [26]. Age-matched lean control NIH male mice maintained on standard rodent maintenance (10% fat, 30% protein and 60% carbohydrate; Trouw Nutrition) were used for comparative purposes throughout. All animal experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act 1986. All animals were housed individually in an air-conditioned room at 22 ± 2°C with a 12 h light:12 h darkness cycle (lights off between 09:30 and 21:30 hours).
Biochemical analysis
All blood samples were taken from the cut tip of the tail vein of conscious mice at the times indicated in the figures and were immediately centrifuged using a Beckman microcentrifuge (Beckman Instruments, Galway, Ireland) for 30 s at 13,000 g. The resulting plasma was then aliquoted into fresh Eppendorf tubes and stored at −20°C prior to glucose and insulin determinations. Plasma glucose was assayed by an automated glucose oxidase procedure using a Beckman Glucose Analyzer II (Beckman Instruments). Plasma and pancreatic insulin were assayed by a modified dextran-coated charcoal radioimmunoassay [27]. Plasma and tissue lipid profile were measured using a Hitachi Automated Analyser 912 (Boehringer Mannheim, Mannheim, Germany).
Statistical analysis
Results are expressed as mean ± SEM. Data were compared using ANOVA, followed by a Student–Newman–Keuls post hoc test. AUC analyses were calculated using the trapezoidal rule with baseline subtraction. p < 0.05 was considered to be statistically significant.
Results
Effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on food intake in normal mice
[d-Leu-4]-OB3 had no significant effect on food intake in normal mice at the doses tested in the current study (Fig. 1a–d). In addition, at a dose of 0.2 nmol/kg (pGlu-Gln)-CCK-8 was devoid of effects on feeding (Fig. 1a). However, (pGlu-Gln)-CCK-8 significantly (p < 0.05 to p < 0.01) reduced food intake at 30, 60 and 90 min post injection at a dose of 1 nmol/kg both alone and in combination with [d-Leu-4]-OB3 (Fig. 1b). At doses of 5 nmol/kg and above, (pGlu-Gln)-CCK-8 significantly (p < 0.05 to p < 0.001) suppressed feeding at all time points compared with saline-treated controls and mice treated with [d-Leu-4]-OB3 (Fig. 1c, d). Combined administration of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 was associated with significant additive effects in suppressing food intake at 120 min post injection (p < 0.05) at a dose of 5 nmol/kg (pGlu-Gln)-CCK-8, and at 60, 90 and 120 min (p < 0.05 to p < 0.01) at a dose of 25 nmol/kg when compared with the same dose of (pGlu-Gln)-CCK-8 administered (Fig. 1c, d).

Dose-dependent effects of (pGlu-Gln)-CCK-8 alone and in combination with [d-Leu-4]-OB3 on food intake in normal mice. (a) White bars, saline controls; black bars, [d-Leu-4]-OB3 (8.33 mg/kg); light grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (0.2 nmol/kg); dark grey bars, (pGlu-Gln)-CCK-8 (0.2 nmol/kg). (b) White bars, saline controls; black bars, [d-Leu-4]-OB3 (8.33 mg/kg); light grey bars, (pGlu-Gln)-CCK-8 (1 nmol/kg); dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (1 nmol/kg). (c) White bars, saline controls; black bars, [d-Leu-4]-OB3 (8.33 mg/kg); light grey bars, (pGlu-Gln)-CCK-8 (5 nmol/kg); dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (5 nmol/kg). (d) White bars, saline controls; black bars, [d-Leu-4]-OB3 (8.33 mg/kg); light grey bars, (pGlu-Gln)-CCK-8 (25 nmol/kg); dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg). Values are mean ± SEM (n = 8). **p < 0.01 and ***p < 0.001 compared with saline control group; † p < 0.05, †† p < 0.01 and ††† p < 0.001 compared with [d-Leu-4]-OB3 group; ‡ p < 0.05 and ‡‡ p < 0.01 compared with combined therapy group
Acute antihyperglycaemic and insulin-releasing activity of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 in normal mice
[d-Leu-4]-OB3 had no significant antihyperglycaemic or insulin-releasing effects when administered in combination with glucose to normal mice (Fig. 2a–f). Administration of 1 nmol/kg (pGlu-Gln)-CCK-8, either alone or in combination with [d-Leu-4]-OB3, also had no effect on plasma glucose or insulin levels following an i.p. glucose load (Fig. 2a, b). However, at a dose of 5 nmol/kg (pGlu-Gln)-CCK-8 significantly (p < 0.001) reduced the overall glycaemic excursion (Fig. 2c). Furthermore, a combination of 5 nmol/kg (pGlu-Gln)-CCK-8 with [d-Leu-4]-OB3 resulted in significantly decreased (p < 0.01) glucose levels 60 min post injection and 0–60 min AUC values (p < 0.01) when compared with (pGlu-Gln)-CCK-8 alone (Fig. 2c). Similar beneficial effects on glucose homeostasis actions were evident at a dose of 25 nmol/kg (pGlu-Gln)-CCK-8 (Fig. 2e). The overall insulin secretory response evoked by (pGlu-Gln)-CCK-8 was significantly (p < 0.001) enhanced when compared with controls at doses of 5 and 25 nmol/kg both alone and in combination with [d-Leu-4]-OB3 (Fig. 2d, f). However, there were no additive insulinotropic effects in the combined treatment group (Fig. 2d, f).

Dose-dependent glucose-lowering (a, c, e) and insulin-releasing (b, d, f) effects of (pGlu-Gln)-CCK-8 alone and in combination with [d-Leu-4]-OB3 in 4 h fasted normal mice. The time of injection is indicated by the arrow (0 min). Plasma glucose and insulin AUC values for 0–60 min post injection are shown in insets. (a, b) Black squares and white bars, saline controls; black triangles and black bars, [d-Leu-4]-OB3 (8.33 mg/kg); open squares and light grey bars, (pGlu-Gln)-CCK-8 (1 nmol/kg); open triangles and dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (1 nmol/kg). (c, d) Black squares and white bars, saline controls; black triangles and black bars, [d-Leu-4]-OB3 (8.33 mg/kg); open squares and light grey bars, (pGlu-Gln)-CCK-8 (5 nmol/kg); open triangles and dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (5 nmol/kg). (e, f) Black squares and white bars, saline controls; black triangles and black bars, [d-Leu-4]-OB3 (8.33 mg/kg); open squares and light grey bars, (pGlu-Gln)-CCK-8 (25 nmol/kg); open triangles and dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg). Values are mean ± SEM for eight mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with glucose alone; † p < 0.05, †† p < 0.01 and ††† p < 0.001 compared with [d-Leu-4]-OB3 group; ‡ p < 0.05, ‡‡ p < 0.01 and ‡‡‡ p < 0.001 compared with (pGlu-Gln)-CCK-8 group
Long-term effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on food intake, body weight and non-fasting plasma glucose and insulin levels in high-fat-fed mice
Administration of (pGlu-Gln)-CCK-8 twice daily for 18 days to high-fat-fed mice resulted in significantly (p < 0.05 to p < 0.001) decreased body weight and accumulated food intake from day 6 onwards (Fig. 3a, b). [d-Leu-4]-OB3 administration had no effect on body weight but there was a slight non-significant trend towards decreased food intake when compared with high-fat-fed controls (Fig. 3a, b). Combined treatment with (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 did not result in any beneficial additive effects on body weight or food intake (Fig. 3a, b). Non-fasting plasma glucose concentrations were restored to lean control levels in all mice treated with (pGlu-Gln)-CCK-8 from day 6 onwards (Fig. 3c). [d-Leu-4]-OB3 therapy was also associated with dramatically reduced (p < 0.05 to p < 0.01) circulating glucose concentrations compared with saline-treated high-fat-fed mice, but these still remained elevated (p < 0.05) compared with lean controls on day 18 (Fig. 3c). No significant changes in plasma insulin levels were noted in mice treated with [d-Leu-4]-OB3 alone (Fig. 3d). (pGlu-Gln)-CCK-8 treatment reduced (p < 0.05) non-fasting insulin levels compared with high-fat-fed controls, but concentrations were still elevated (p < 0.05) compared with lean controls on day 18 (Fig. 3d). Combined (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 administration returned non-fasting insulin to lean control levels from day 12 onwards (Fig. 3d).

Effects of twice daily (pGlu-Gln)-CCK-8, [d-Leu-4]-OB3 or combined peptide administration on body weight (a), accumulated food intake (b), plasma glucose (c) and plasma insulin (d) in high-fat-fed mice. Variables were measured for 6 days prior to and 18 days during (indicated by horizontal black bar) treatment. (a, c, d) Black squares, high-fat-fed controls; open triangles, [d-Leu-4]-OB3 (8.33 mg/kg); open inverted triangles, (pGlu-Gln)-CCK-8 (25 nmol/kg); black diamonds, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg); open squares, lean controls. (b) White bars, high-fat-fed controls; black bars, [d-Leu-4]-OB3 (8.33 mg/kg); light grey bars, (pGlu-Gln)-CCK-8 (25 nmol/kg); dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg); striped bars, lean controls. Values are mean ± SEM for eight mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with high-fat group; † p < 0.05, †† p < 0.01 and ††† p < 0.001 compared with [d-Leu-4]-OB3 group; ‡ p < 0.05, ‡‡ p < 0.01 and ‡‡‡ p < 0.001 compared with (pGlu-Gln)-CCK-8 group; § p < 0.05 and §§§ p < 0.001 compared with combined therapy group
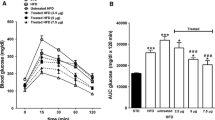
Long-term effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on glucose tolerance and insulin sensitivity in high-fat-fed mice
Plasma glucose levels in glucose tolerance tests were significantly reduced (p < 0.001) at 15 min in all treatment groups compared with high-fat-fed controls (Fig. 4a). This was corroborated by significantly (p < 0.05) decreased 0–60 min AUC values (Fig. 4b). However, the overall glycaemic excursion was still significantly (p < 0.01 to p < 0.001) elevated in all treatment groups compared with lean control mice (Fig. 4b). Glucose-stimulated plasma insulin concentrations were significantly reduced 30 min post injection in mice treated with (pGlu-Gln)-CCK-8 alone or in combination with [d-Leu-4]-OB3 compared with high-fat-fed mice treated with either saline (p < 0.01) or [d-Leu-4]-OB3 (p < 0.05) (Fig. 4c). AUC analysis revealed similar effects with all treatment groups evoking significantly (p < 0.05 to p < 0.001) reduced overall insulin secretory responses which remained elevated (p < 0.001) compared with lean controls (Fig. 4d). As shown in Fig. 4e, f, the hypoglycaemic action of insulin was significantly augmented 30 min post injection (p < 0.001), with lower AUC values (p < 0.001) in high-fat-fed mice treated with (pGlu-Gln)-CCK-8 alone or in combination with [d-Leu-4]-OB3. Moreover, insulin sensitivity was not significantly different from that of lean control levels in these mice (Fig. 4e, f). [d-Leu-4]-OB3 treatment alone resulted in significantly (p < 0.001) improved insulin sensitivity in terms of overall AUC measures compared with high-fat-fed controls, but was still significantly (p < 0.001) inferior to that of mice treated with (pGlu-Gln)-CCK-8 alone or in combination with [d-Leu-4]-OB3 (Fig. 4e, f).

Effects of twice daily (pGlu-Gln)-CCK-8, [d-Leu-4]-OB3 or combined peptide administration on glucose tolerance and plasma insulin response to glucose (a–d) and insulin sensitivity (e, f) in high-fat-fed mice. Tests were conducted after twice daily treatment for 18 days. (a–f) Black squares and white bars, high-fat-fed controls; open triangles and black bars, [d-Leu-4]-OB3 (8.33 mg/kg); open inverted triangles and light grey bars, (pGlu-Gln)-CCK-8 (25 nmol/kg); black diamonds and dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg); open squares and striped bars, lean controls. Values are mean ± SEM for eight mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with high-fat-fed group; † p < 0.05, †† p < 0.01 and ††† p < 0.001 compared with [d-Leu-4]-OB3 group; ‡‡ p < 0.01 and ‡‡‡ p < 0.001 compared with (pGlu-Gln)-CCK-8 group; §§§ p < 0.001 compared with combined therapy group. AAC, area above the curve
Long-term effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on blood lipid profile, locomotor activity and indirect calorimetry in high-fat-fed mice
Circulating triacylglycerol levels were significantly (p < 0.01) reduced compared with high-fat-fed mice in all treatment groups, with values similar to those of lean controls (Fig. 5a). LDL-cholesterol concentrations were unaffected by [d-Leu-4]-OB3 treatment, but were significantly (p < 0.001) lowered in mice treated with (pGlu-Gln)-CCK-8 (Fig. 5b). In addition, combined (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 therapy returned LDL-cholesterol to that of lean control levels (Fig. 5b). Circulating HDL-cholesterol was unaffected by high-fat feeding or any of the treatment regimens (Fig. 5c).

Effects of twice daily (pGlu-Gln)-CCK-8, [d-Leu-4]-OB3 or combined peptide administration on blood lipid profile (a–c) and locomotor activity (d, e) in high-fat-fed mice. Variables were measured after twice daily treatment for 18 days. (a–e) White bars, high-fat-fed controls; black bars, [d-Leu-4]-OB3 (8.33 mg/kg); light grey bars, (pGlu-Gln)-CCK-8 (25 nmol/kg); dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg); striped bars, lean controls. Values are mean ± SEM for eight mice. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with high-fat-fed group; † p < 0.05 and ††† p < 0.001 compared with [d-Leu-4]-OB3 group; ‡ p < 0.05 and ‡‡‡ p < 0.001 compared with (pGlu-Gln)-CCK-8 group; §§ p < 0.01 and §§§ p < 0.001 compared with combined therapy group
Figure 5d, e depicts the effects of 18 days of twice daily treatment with (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on locomotor activity. Both peptides significantly (p < 0.05 to p < 0.01) reduced ambulatory activity in high-fat-fed mice as assessed by X-beam breaks during the light phase (Fig. 5d). Surprisingly, combined therapy was not associated with altered X-beam breaks during the light phase (Fig. 5d). Rearing or jumping episodes during the light phase, as assessed by Z-beam breaks, were significantly (p < 0.001) decreased in all mice treated with (pGlu-Gln)-CCK-8 compared with mice treated with [d-Leu-4]-OB3 alone and high-fat-fed controls (Fig. 5d). All high-fat-fed mice had increased (p < 0.05 to p < 0.01) ambulatory activity during the dark phase when compared with lean controls (Fig. 5e). Interestingly, mice treated with (pGlu-Gln)-CCK-8 had significantly increased X- and Z-beam breaks compared with mice treated with [d-Leu-4]-OB3 (p < 0.05 and p < 0.001, respectively) and significantly (p < 0.001) increased Z-beam breaks compared with high-fat-fed controls during the dark phase (Fig. 5d). Administration of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 had no significant effect on O2 consumption, CO2 production and RER when compared with high-fat-fed controls (Fig. 6a–c). However, energy expenditure was significantly (p < 0.01 to p < 0.001) decreased in all mice treated with (pGlu-Gln)-CCK-8 compared with high-fat-fed controls and [d-Leu-4]-OB3-treated mice (Fig. 6d). All aspects of indirect calorimetry were significantly (p < 0.001) different compared with lean control mice in all high-fat treatment groups (Fig. 6a–d).

Effects of twice daily (pGlu-Gln)-CCK-8, [d-Leu-4]-OB3 or combined peptide administration on O2 consumption (a), RER (b), CO2 production (c) and energy expenditure (d) in high-fat-fed mice. Variables were measured after twice daily treatment for 18 days. The dark phase is indicated by the black horizontal bar. Insets depict the consequence of combining light and dark phase data. (a–f) Black squares and white bars, high-fat-fed controls; open triangles and black bars, [d-Leu-4]-OB3 (8.33 mg/kg); open inverted triangles and light grey bars, (pGlu-Gln)-CCK-8 (25 nmol/kg); black diamonds and dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg); open squares and striped bars, lean controls. Values are mean ± SEM for eight mice. ***p < 0.001 compared with high-fat-fed group; †† p < 0.01 and ††† p < 0.001 compared with [d-Leu-4]-OB3 group; ‡‡‡ p < 0.001 compared with (pGlu-Gln)-CCK-8 group; §§§ p < 0.001 compared with combined therapy group
Long-term effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on tissue triacylglycerol content and pancreatic insulin levels in high-fat-fed mice
Triacylglycerol content of adipose and liver tissue was significantly (p < 0.001) decreased in mice treated with (pGlu-Gln)-CCK-8 alone or in combination with [d-Leu-4]-OB3 when compared with high-fat-fed controls (Fig. 7a, b). Liver triacylglycerol content of these mice was retuned to lean control levels (Fig. 7b). Combined (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 treatment significantly (p < 0.05) reduced muscle triacylglycerol concentrations compared with saline-treated high-fat-fed controls (Fig. 7c). There was no significant difference in pancreatic insulin content of all treatment groups when compared with high-fat-fed control mice (Fig. 7d).

Effects of twice daily (pGlu-Gln)-CCK-8, [d-Leu-4]-OB3 or combined peptide administration on tissue lipid profile (a–c) and pancreatic insulin content (d) in high-fat-fed mice. Variables were measured after twice daily treatment for 18 days. (a–e) White bars, high-fat-fed controls; black bars, [d-Leu-4]-OB3 (8.33 mg/kg); light grey bars, (pGlu-Gln)-CCK-8 (25 nmol/kg); dark grey bars, [d-Leu-4]-OB3 (8.33 mg/kg) + (pGlu-Gln)-CCK-8 (25 nmol/kg); striped bars, lean controls. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with high-fat-fed group; ††† p < 0.001 compared with [d-Leu-4]-OB3 group; ‡‡‡ p < 0.001 compared with (pGlu-Gln)-CCK-8 group; §§§ p < 0.001 compared with combined therapy group
Discussion
CCK and leptin are two important regulators of food intake and overall energy balance that have significant, but as yet untapped, therapeutic potential. In this regard, structure–function analysis has shown that the amino acid sequences of CCK and leptin can be dissociated into distinct shorter fragments that still retain biological actions of the parent peptides [7, 12]. Thus, development of shorter bioactive forms of CCK and leptin might reduce production costs and facilitate non-injectable drug delivery, thereby increasing their overall utility. Therefore, the present study has evaluated the bioactivity and possible therapeutic potential of fragment CCK and leptin peptides, namely (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3.
We and others have recently highlighted the involvement of CCK receptor signalling in insulin secretion and for controlling glucose homeostasis both in normal rodents and in animal models with established obesity/diabetes [7, 28]. However, leptin is generally considered to inhibit insulin secretion [29]. In the current study, we were unable to reveal any effect of [d-Leu-4]-OB3 on in vivo insulin secretion or glucose homeostasis. However, the leptin fragment did significantly augment the glucose-lowering actions of (pGlu-Gln)-CCK-8. By contrast, the full 167 amino acid leptin peptide has previously been shown to inhibit the insulinotropic response of CCK-8 in the perfused rat pancreas [30]. This difference might reflect the artificial nature of the in vitro system. Further study into the additive effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on glucose homeostasis and endocrine pancreatic secretions is required. For example, measurement of C-peptide levels could help determine whether increased insulin secretion or decreased liver clearance is involved. By contrast, the synergistic interaction between leptin and CCK to reduce short-term food intake is well established [31–33]. Thus, as expected, in the current study combined administration of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 exhibited significantly enhanced acute feeding-suppressive effects compared with either peptide alone.
In order fully to appreciate potential additive beneficial effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 we employed a chronic treatment regimen in high-fat-fed mice. Unlike the acute effects, the longer-term beneficial actions of (pGlu-Gln)-CCK-8 on inhibiting energy intake were not augmented by [d-Leu-4]-OB3, which was translated to similar observations on body weight regulation. Indeed, the lack of synergistic effects on body weight control mirrors the increased difficulty in losing weight as observed in clinical and experimental presentations of diabetes. However, a previous report has detailed that additive effects of leptin and CCK on body weight reduction were only observed with intracerebroventricular (i.c.v.), and not i.p., peptide administration [18]. Nonetheless, in harmony with previous observations [7], sub-chronic administration of (pGlu-Gln)-CCK-8 twice daily to high-fat-fed mice resulted in a marked improvement in metabolic status, glucose homeostasis and insulin sensitivity, indicative of the increased metabolic efficiency that typically accompanies weight loss. Importantly, we have previously shown that (pGlu-Gln)-CCK-8 does not affect insulin sensitivity when administered acutely to mice [7]. Decreased energy intake was also observed together with decreases in body weight gain, circulating insulin and triacylglycerol levels, as well as reduced accumulation of tissue triacylglycerol. Similar beneficial effects were not noted with [d-Leu-4]-OB3 treatment despite previous reports that [d-Leu-4]-OB3 reduced energy intake and body weight gain in C57BL/6J ob/ob mice following prolonged administration [12]. These differences may be a reflection of a variation in dosing regimen, length of experimentation or the different strains of mice used. Moreover, it has been reported that high-fat feeding attenuates the response to leptin in rodents [34] and induces leptin resistance under certain circumstances [35]. Furthermore, sub-chronic administration of [d-Leu-4]-OB3 could potentially lead to the generation of leptin antibodies, although this seems unlikely given the minor structural differences between [d-Leu-4]-OB3 and the native leptin fragment. Moreover, previous clinical studies with leptin have noted non-neutralising effects of antibodies generated against leptin [36, 37], in parallel with other observations using similar regulatory peptides [38]. In addition, the prominent beneficial effects of (pGlu-Gln)-CCK-8 on glucose tolerance, insulin sensitivity and triacylglycerol content of adipose and liver tissue were not enhanced by concurrent administration of [d-Leu-4]-OB3. This might reflect the good efficacy of (pGlu-Gln)-CCK-8 alone and the relatively high doses of peptides employed, which could preclude an additive action in respect to these variables. It may be that dose adjustments could yield more synergistic effects during sub-chronic testing, but such assessments were not possible in the current study. In addition, beneficial effects could be strongly associated with body weight reductions, which were prominent in all (pGlu-Gln)-CCK-8-treated mice.
Development of drug tolerance has been a major issue in the realisation CCK-8-based therapies [39, 40]. Moreover, recently it has been shown that leptin resistance within vagal afferents reduces the sensitivity to CCK receptor-mediated effects [22]. Furthermore, the beneficial effects of (pGlu-Gln)-CCK-8 were less obvious in leptin-deficient ob/ob mice than in high-fat-fed mice [7]. Combined administration of pharmacological doses of leptin and CCK therapy, as in the current study, may help to overcome this. Indeed, we noted prominent additive effects of 18 days of twice daily treatment with (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on circulating glucose, insulin and LDL-cholesterol levels as well as on the triacylglycerol content of muscle tissue. It could be possible that the combined treatment regimen resulted in increased leptin responsiveness in these animals; further studies, however, are needed to confirm this. Thus, high-fat feeding can be associated with leptin resistance [35]. Nonetheless, the acute synergistic effects of (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3 on inhibiting food intake were much more pronounced at elevated drug concentrations in normal mice. As such, there are a number of postulated mechanisms for the cooperative actions between CCK and leptin, all of which concern action at the level of the vagal afferents. Thus, CCK and leptin have synergistic effects to increase cytosolic calcium concentrations and activate vagal afferent neurons [2, 41], leading to the beneficial effects on the regulation of energy balance [33]. Further to this, there is now evidence that leptin–CCK interactions extend beyond neuronal activation and include changes in gene expression. Leptin is thought to stimulate expression of the early growth response factor-1 gene and CCK promotes its translocation to the nucleus [32], which in turn leads to regulation of expression of the satiety peptide cocaine- and amphetamine-regulated transcript [42]. Moreover, CCK has been shown to facilitate the biological action of leptin and increase the permeability of the blood–brain barrier to leptin [17–19]. Further to this, we have recently shown that modulation of neuropeptide Y- and melanocortin-related pathways may be involved in the beneficial effects of (pGlu-Gln)-CCK-8 therapy in high-fat-fed mice [43]. Interestingly, CCK was also recently shown to have possible synergistic effects with GLP-1 in terms of the central regulation of food intake and energy balance [44], highlighting the plasticity of pathways involved in energy balance [42].
In order to further clarify the mechanism behind the observed beneficial effects in the current study, we assessed aspects of indirect calorimetry following 18 days' treatment with (pGlu-Gln)-CCK-8, [d-Leu-4]-OB3 or a combination of both peptides. Although extensive studies have been conducted on the additive effects of leptin and CCK on energy intake [4], there is much less information on their possible combined effects on locomotor activity and energy expenditure. Explorative episodes (Z-beam breaks) were elevated during the dark phase but reduced during the light phase in all mice treated with (pGlu-Gln)-CCK-8, with no overall net effect. This is encouraging given that CCK receptor activation has been associated with behavioural changes in both rodents and humans [45, 46]. Moreover, we have previously confirmed that sub-chronic twice daily administration of 25 nmol/kg (pGlu-Gln)-CCK-8 was not associated with any detrimental effects on behaviour in high-fat-fed mice [7]. Interestingly, locomotor activity was decreased in the light phase by both (pGlu-Gln)-CCK-8 and [d-Leu-4]-OB3, but not by combined treatment, which is intriguing and merits further investigation. Energy expenditure was also decreased by (pGlu-Gln)-CCK-8, which could be a direct reflection of decreased activity [47]. Nonetheless, given the prominent effects of (pGlu-Gln)-CCK-8 on body weight reduction, elevations of energy expenditure might have been predicted, although measurements were made on day 18 when body weight regulation appears to have stabilised. Furthermore, unlike native leptin, [d-Leu-4]-OB3 does not possess effects on thermogenesis [48], which may also partly explain the above observations.
In conclusion, the present study indicates that once daily injection of either [d-Leu-4]-OB3, or particularly (pGlu-Gln)-CCK-8, represents an effective means of improving metabolic control in obesity/diabetes. Overall our data indicate that there was also evidence of beneficial synergistic effects, which requires further physiological study as a novel therapeutic approach for patients with obesity-driven forms of diabetes.
Abbreviations
- CCK:
-
Cholecystokinin
- NIH:
-
National Institutes of Health
- RER:
-
Respiratory exchange ratio
References
Murphy KG, Bloom SR (2006) Gut hormones and the regulation of energy homeostasis. Nature 444:854–859
Peters JH, Simasko SM, Ritter RC (2006) Modulation of vagal afferent excitation and reduction of food intake by leptin and cholecystokinin. Physiol Behav 89:477–485
Brenner L, Yox DP, Ritter RC (1993) Suppression of sham feeding by intraintestinal nutrients is not correlated with plasma cholecystokinin elevation. Am J Physiol 264:R972–R976
Dockray GJ, Burdyga G (2011) Plasticity in vagal afferent neurones during feeding and fasting: mechanisms and significance. Acta Physiol (Oxf) 201:313–321
Baldwin BA, Parrott RF, Ebenezer IS (1998) Food for thought: a critique on the hypothesis that endogenous cholecystokinin acts as a physiological satiety factor. Prog Neurobiol 55:477–507
Rehfeld JF, Friis-Hansen L, Goetze JP, Hansen TV (2007) The biology of cholecystokinin and gastrin peptides. Curr Top Med Chem 7:1154–1165
Irwin N, Frizelle P, Montgomery IA, Moffett RC, O’Harte FPM, Flatt PR (2012) Beneficial effects of the novel cholecystokinin agonist (pGlu-Gln)-CCK-8 in animal models of obesity/diabetes. Diabetologia 55:2747–2258
Pénicaud L, Meillon S, Brondel L (2012) Leptin and the central control of feeding behavior. Biochimie 94:2069–2074
Benoit SC, Clegg DJ, Seeley RJ, Woods SC (2004) Insulin and leptin as adiposity signals. Recent Prog Horm Res 59:267–285
Pelleymounter MA, Cullen MJ, Baker MB et al (1995) Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269:540–543
Gonzalez LC, Pinilla L, Tena-Sempere M, Aguilar E (1999) Leptin(116–130) stimulates prolactin and luteinizing hormone secretion in fasted adult male rats. Neuroendocrinology 70:213–220
Grasso P, Rozhavskaya-Arena M, Leinung MC, Lee DW (2001) [D-LEU-4]-OB3, a synthetic leptin agonist, improves hyperglycemic control in C57BL/6J ob/ob mice. Regul Pept 101:123–129
Waldrop MA, Leinung MC, Lee DW, Grasso P (2010) Intranasal delivery of mouse [d-Leu-4]-OB3, a synthetic peptide amide with leptin-like activity, improves energy balance, glycaemic control, insulin sensitivity and bone formation in leptin-resistant C57BLK/6-m db/db mice. Diabetes Obes Metab 12:871–875
Gorska E, Popko K, Stelmaszczyk-Emmel A, Ciepiela O, Kucharska A, Wasik M (2010) Leptin receptors. Eur J Med Res 15:50–54
Sahu A (2004) A hypothalamic role in energy balance with special emphasis on leptin. Endocrinol 145:2613–2620
Burdyga G, Spiller D, Morris R et al (2002) Expression of the leptin receptor in rat and human nodose ganglion neurones. Neuroscience 109:339–347
Matson CA, Reid DF, Cannon TA, Ritter RC (2000) Cholecystokinin and leptin act synergistically to reduce body weight. Am J Physiol Regul Integr Comp Physiol 278:R882–R890
Merino B, Cano V, Guzmán R, Somoza B, Ruiz-Gayo M (2008) Leptin-mediated hypothalamic pathway of cholecystokinin (CCK-8) to regulate body weight in free-feeding rats. Endocrinology 149:1994–2000
Cano V, Merino B, Ezquerra L, Somoza B, Ruiz-Gayo M (2008) A cholecystokinin-1 receptor agonist (CCK-8) mediates increased permeability of brain barriers to leptin. Br J Pharmacol 154:1009–1015
Bado A, Levasseur S, Attoub S et al (1998) The stomach is a source of leptin. Nature 394:790–793
Guilmeau S, Buyse M, Tsocas A, Laigneau JP, Bado A (2003) Duodenal leptin stimulates cholecystokinin secretion: evidence of a positive leptin-cholecystokinin feedback loop. Diabetes 52:1664–1672
de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE (2012) Leptin resistance in vagal afferent neurons inhibits cholecystokinin signaling and satiation in diet induced obese rats. PLoS One 7:e32967
Kerr BD, Irwin N, O'Harte FP, Bailey CJ, Flatt PR, Gault VA (2009) Fatty acid derivatised analogues of glucose-dependent insulinotropic polypeptide with improved antihyperglycaemic and insulinotropic properties. Biochem Pharmacol 78:1008–1016
Montgomery IA, Irwin N, Flatt PR (2010) Active immunization against (Pro3)GIP improves metabolic status in high-fat-fed mice. Diabetes Obes Metab 12:744–751
Friedewald WT, Levy RI, Fredrickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 18:499–502
Gault VA, Porter DW, Irwin N, Flatt PR (2011) Comparison of sub-chronic metabolic effects of stable forms of naturally occurring GIP(1–30) and GIP(1–42) in high-fat fed mice. J Endocrinol 208:265–271
Flatt PR, Bailey CJ (1981) Abnormal plasma glucose and insulin responses in heterozygous lean (ob/+) mice. Diabetologia 20:573–577
Lo CM, Obici S, Dong HH et al (2011) Impaired insulin secretion and enhanced insulin sensitivity in cholecystokinin-deficient mice. Diabetes 60:2000–2007
Zhao AZ, Bornfeldt KE, Beavo JA (1998) Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J Clin Invest 102:869–873
Silvestre RA, Rodríguez-Gallardo J, Egido EM, Marco J (2001) Effect of leptin on insulin, glucagon and somatostatin secretion in the perfused rat pancreas. Horm Metab Res 33:207–212
Barrachina MD, Martínez V, Wang L, Wei JY, Taché Y (1997) Synergistic interaction between leptin and cholecystokinin to reduce short-term food intake in lean mice. PNAS 94:10455–10460
de Lartigue G, Lur G, Dimaline R, Varro A, Raybould H, Dockray GJ (2010) EGR1 is a target for cooperative interactions between cholecystokinin and leptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology 151:3589–3599
Wang L, Barachina MD, Martínez V, Wei JY, Taché Y (2000) Synergistic interaction between CCK and leptin to regulate food intake. Regul Pept 92:79–85
Lin L, Martin R, Schaffhauser AO, York DA (2001) Acute changes in the response to peripheral leptin with alteration in the diet composition. Am J Physiol Regul Integr Comp Physiol 280:R504–R509
Harris RB, Bowen HM, Mitchell TD (2003) Leptin resistance in mice is determined by gender and duration of exposure to high-fat diet. Physiol Behav 78:543–555
Moon HS, Matarese G, Brennan AM et al (2011) Efficacy of metreleptin in obese patients with type 2 diabetes: cellular and molecular pathways underlying leptin tolerance. Diabetes 60:1647–1656
Ebihara K, Kusakabe T, Hirata M et al (2007) Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab 92:532–541
Fineman MS, Mace KF, Diamant M et al (2012) Clinical relevance of anti-exenatide antibodies: safety, efficacy and cross-reactivity with long-term treatment. Diabetes Obes Metab 14:546–554
Crawley JN, Beinfeld MC (1983) Rapid development of tolerance to the behavioural actions of cholecystokinin. Nature 302:703–706
Mineka S, Snowdon CT (1978) Inconsistency and possible habituation of CCK-induced satiety. Physiol Behav 21:65–72
Peters JH, Karpiel AB, Ritter RC, Simasko SM (2004) Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology 145:3652–3657
Dockray GJ (2009) Cholecystokinin and gut-brain signalling. Regul Pept 155:6–10
Montgomery IA, Irwin N, Flatt PR (2013) Beneficial effects of (pGlu-Gln)-CCK-8 on energy intake and metabolism in high fat fed mice are associated with alterations of hypothalamic gene expression. Horm Metab Res. doi:10.1055/s-0032-1331767
Hisadome K, Reimann F, Gribble FM, Trapp S (2011) CCK stimulation of GLP-1 neurons involves α1-adrenoceptor-mediated increase in glutamatergic synaptic inputs. Diabetes 60:2701–2709
Bradwejn J (1993) Neurobiological investigations into the role of cholecystokinin in panic disorder. J Psychiatry Neurosci 18:178–188
Wang H, Wong PT, Spiess J, Zhu YZ (2005) Cholecystokinin-2 (CCK2) receptor-mediated anxiety-like behaviors in rats. Neurosci Biobehav Rev 29:1361–1373
Porter WD, Flatt PR, Hölscher C, Gault VA (2012) Liraglutide improves hippocampal synaptic plasticity associated with increased expression of Mash1 in ob/ob mice. Int J Obes (Lond). doi:10.1038/ijo.2012.91
Rozhavskaya-Arena M, Lee DW, Leinung MC, Grasso P (2000) Design of a synthetic leptin agonist: effects on energy balance, glucose homeostasis, and thermoregulation. Endocrinology 141:2501–2507
Funding
These studies were supported by the SAAD Trading and Contracting Company and the Department of Education and Learning, Northern Ireland, UK.
Duality of interest
N. Irwin and P.R. Flatt hold shares with Diabetica Ltd, which has patents for exploitation of peptide therapeutics.
Contribution statement
NI conceived the study, participated in the analysis and interpretation of data, drafted the manuscript and revised it critically for intellectual content. IAM participated in the analysis and interpretation of data and drafted the manuscript and revised it critically for intellectual content. PRF conceived the study, drafted the manuscript and revised it critically for intellectual content. All authors approved the final version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Irwin, N., Montgomery, I.A. & Flatt, P.R. Comparison of the metabolic effects of sustained CCK1 receptor activation alone and in combination with upregulated leptin signalling in high-fat-fed mice. Diabetologia 56, 1425–1435 (2013). https://doi.org/10.1007/s00125-013-2878-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2878-0