
Abstract
Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine involved in many inflammatory reactions and disorders, and it has become evident that it also affects glucose homeostasis. The protein is produced by pancreatic beta cells and can promote the release of insulin. It also modulates glucose uptake, glycolysis and insulin resistance in insulin target cells such as the adipocyte, myocyte and cardiomyocyte. Possessing both immunological and endocrinological properties, MIF has been associated with the development of type 1 and type 2 diabetes, and it may be important in the setting of islet transplantation. The present review summarises our current knowledge, based on clinical and research data, on the impact of MIF on both physiological and pathological aspects of glucose metabolism.
Similar content being viewed by others

Macrophage migration inhibitory factor (MIF) was one of the first of the proinflammatory cytokines to be described. In the 1960s it was reported to inhibit the migration of macrophages in vitro [1, 2], with a maximum effect achieved at 80 pmol/l [3]. Its broad proinflammatory activities were more extensively described during the 1990s, following the elucidation of the DNA sequence for the protein (chromosome 22 in humans and chromosome 10 in mice), which led to the production of the recombinant protein [4–7].
Mif is known to be constitutively expressed in numerous types of tissue, such as lung, skin, the gastrointestinal and urinary tracts, several endocrine glands (pancreatic beta cells, ovary, testis, hypothalamus, adrenal and pituitary glands) and cells of the immune system (T and B cells, monocytes/macrophages, neutrophils, eosinophils, basophils, mast cells, dentritic cells) [8–13]. In addition, it is involved in various acute and chronic inflammatory diseases, including sepsis, adult respiratory distress syndrome, glomerulonephritis, arthritis, colitis and gastritis [14]. There is a close relationship between the role of MIF in the inflammatory/immune response and its role in glucose metabolism. Acute inflammation can lead to abnormal glucose regulation, while type 1 and type 2 diabetes are associated with chronic immune and inflammatory reactions [15].
Increasing evidence suggests that MIF plays a key role in glucose homeostasis during periods of stress and in the development of type 1 and type 2 diabetes. The present review describes the role of MIF in the immune response and its physiological and pathological effects on glucose metabolism, using both research and clinical data. In addition, potential sites for therapeutic regulation of MIF activity are explored.
Structural and signalling aspects of MIF
The normal range for plasma MIF is 0.2–0.5 nmol/l in humans [16]. The X-ray crystal structure of the protein, at a resolution of 2.6 Å [17], shows that this small (12 kDa) hydrophilic cytokine is composed of a trimer of identical subunits. The MIF receptor has been identified as the CD74–CD44 complex, which can be found on most nucleated cells [18–22], and the cytokine binds primarily to the extracellular transmembrane component, CD74. The CD44 component is required for the transduction of binding signals mediated by the p44/p42 mitogen-activated protein kinase (extracellular signal-regulated protein kinase 1/2) pathway [18, 22]. It should be noted that CD74 is also known as the MHC class II invariant chain, while CD44 is a recyclable receptor with an important role in cell–extracellular matrix interactions [23].
MIF and innate and adaptive immunity
The proinflammatory effect of MIF in the innate immune response has been extensively studied (see text box: Impact of MIF on innate and adaptive immunity and glucose metabolism, Fig. 1a). Endotoxin and various inflammatory cytokines, including TNF-α and IFN-γ (but not IL-1β and IL-6), increase Mif expression and the release of the protein by macrophages, while IL-10 inhibits its release [10, 24]. MIF activates macrophages in an autocrine and paracrine manner, generating a positive-feedback loop. It promotes the production of TNF-α, cyclooxygenase 2 and prostaglandin E2, amplifying the inflammatory reaction [5, 10, 14, 25]. MIF also enhances the production of TNF-α and type 1 IL-1 receptors on various cells, as well as the production of Toll-like receptor 4 on macrophages [26–28].


Schematic diagrams of the known effect of MIF on innate (a) and adaptive (b) immunity. COX2, cyclooxygenase 2; IL-1R, IL-1 receptor; TNF-αR, TNF-α receptor
When MIF has been released at a site of inflammation, it promotes the recruitment of more leucocytes, thus increasing the innate response and propagating an adaptive response. It increases the expression of the adhesion molecules vascular cell adhesion molecule-1 and intercellular adhesion molecule-1, which mediates monocyte adherence to the vascular endothelium located near the site of inflammation [29]. MIF binds to the chemokine receptors CXCR2 and CXCR4, thereby promoting monocyte and lymphocyte arrest in the area of inflammation [30]. MIF contributes to leucocyte transmigration from the vessels to the inflamed tissue by promoting endothelial cell production of monocyte chemotaxis protein-1 (MCP-1), a potent chemoattractant [31]. During the inflammatory response, the turnover of immune cells is elevated, in part due to activation-induced cell death (apoptosis). MIF maintains the inflammatory response by decreasing p53-mediated lymphocyte apoptosis and improving macrophage survival [25, 32].
Several recent articles suggest that MIF plays a significant role in the adaptive immune response (Fig. 1b). MIF is secreted by T lymphocytes under normal conditions and is increased in response to various proteins, including anti-CD3, phorbol 12-myristate 13-acetate (PMA) and concanavalin A, all of which are potent in vitro lymphocyte stimulators [33, 34]. Similar to its effects on macrophages during an innate response, MIF has an autocrine effect on lymphocytes. It promotes the release of IL-2 and TNF-α, the expression of the gene encoding the IL-2 receptor (CD25), and, ultimately, the activation and proliferation of T and B cells [33, 35, 36]. MIF affects the balance between T helper cell type 1 (Th1) and Th2 responses and alters the type of immunoglobulin secreted by B cells [37–39]. However, the impact of MIF on the Th1 vs Th2 pathway appears to be dependent upon the disease process involved [37–39]. It remains unclear whether MIF modulates the secretion of immunoglobulin directly or indirectly through mediators. MIF also contributes to cytotoxic activity against tumour cells [34] and is involved in the delayed-type hypersensitivity reaction [40–42].
MIF and glucose metabolism
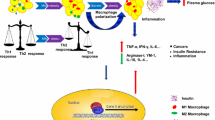
Besides its known role in the immune response, MIF influences glucose metabolism at several levels, affecting both insulin production in the pancreatic beta cell and the cells targeted by insulin (see text box: Impact of MIF on innate and adaptive immunity and glucose metabolism, Fig. 2) [15]. MIF is produced in beta cells and is co-localised with insulin in the secretory granules [15]. The expression of MIF within the beta cell and its plasma levels follow a circadian rhythm, with higher levels reported during the day [16, 43]. Its production is regulated by glucose in a time- and concentration-dependent manner [44, 45]. Once released, MIF has a positive autocrine action on insulin secretion, which ultimately leads to lower levels of glucose and MIF [44]. These observations are based on the increased glucose-induced insulin release from rodent islets in the presence of recombinant MIF in vitro [44]. In addition, the use of anti-MIF neutralising antibodies or the downregulation of Mif expression decreases glucose-induced insulin secretion in perifusion studies performed with rat islets or an insulin-producing cell line (INS-1) [44].

Schematic diagram of the known impact of MIF on glucose metabolism
MIF is also produced by cells targeted by insulin, including myocytes, cardiomyocytes and adipocytes, both constitutively and in response to stimuli such as TNF-α [46–52]. Its release from adipocytes appears to be site-specific, as adipocytes from the subcutaneous tissue and the omentum secrete approximately ten times more MIF than mammary adipocytes [50]. MIF has an autocrine/paracrine effect on adipocytes, and has been suggested to mediate the effects of TNF-α on glucose catabolism, specifically in the context of stress [51]. MIF also induces enhanced glucose uptake and glycolysis in muscle, as assessed by higher levels of the enzyme 6-phosphofructo-2-kinase (PFK-2), which is associated with an increase in the synthesis of fructose 2,6-biphosphate, a powerful positive allosteric regulator of glycolysis, further leading to enhanced lactate production [49]. Experiments on mouse heart metabolism have suggested that MIF exerts its metabolic effects at least in part via the AMP-activated protein kinase pathway [52], which acts as a sensor of the energy state of the cell and induces diverse signals to increase ATP production. This can result in a rise in glucose uptake following increases in the translocation of GLUT4 to the cell surface and PFK-2 activity, leading to a higher rate of glycolysis [52]. As a consequence of the MIF-mediated metabolic effects, and in contrast to wild-type controls, MIF knockout (KO) animals or mice treated with anti-MIF neutralising antibody exhibit normal blood glucose levels, lactate response and liver glycogen content following the administration of endotoxin or TNF-α [49, 51]. In addition, MIF KO animals or mice treated with neutralising anti-MIF antibody demonstrate a selective increase in insulin-mediated glucose uptake into white adipose tissue after TNF-α treatment, while glucose uptake in skeletal muscle and brown adipose tissue and hepatic glucose production are not affected [51]. MIF is a necessary mediator of TNF-α inhibition of the insulin signal transduction leading to insulin resistance [51, 53]. This effect is related to reduced phosphorylation of the protein kinase Akt, which together with a phosphatidylinositol 3-kinase, is necessary for phosphorylation of IRS-1, a secondary messenger of the insulin receptor involved in stimulating the transcription of insulin-regulated genes [51].
MIF and stress
MIF has been extensively studied as a mediator of the response to stress in humans [54]. Very high serum levels of MIF (up to tenfold above normal range) can be found in patients suffering from various insults, including major surgery (liver resection), systemic inflammatory response syndrome, severe sepsis or septic shock. Higher levels are also seen in patients who ultimately die [55–57].
Several animal models have further confirmed the role of MIF in the setting of stress [58]. MIF is produced at all levels of the hypothalamic–pituitary–adrenal axis, but pituitary production is the most striking [59]. In the pituitary gland, MIF is located in the secretory granules of the corticotropic cells, which also contain adrenocorticotropic hormone (ACTH) and thyroid-stimulating hormone [60, 61]. The expression of Mif and release of the protein are induced by corticotropin-releasing hormone [60, 62], and MIF levels are inversely correlated with the adrenal response, as reflected by low levels of serum cortisol and increased levels of ACTH following MIF release [57]. In contrast, cortisol induces MIF production from various tissues, including the adrenal gland [63].
In mouse models of septic shock, intracellular stores of MIF are released from various tissues (pituitary, adrenal, lung, liver, spleen, kidney and skin) within a few hours, with the Mif transcriptional response seen later [59]. Following such an event, the pituitary content of MIF is decreased. MIF appears to play a central role in stress, as its administration leads to an increased mortality rate, whereas anti-MIF neutralising antibodies or the absence of MIF can protect mice from shock and death [64, 65].
Stress is typically characterised by an increase in the cellular uptake of glucose and an increase in the production of glucose by the liver. During the first phase, the increased insulin resistance leads to some degree of hyperglycaemia [66]. However, hypoglycaemia subsequently occurs, probably as a result of an inappropriately high level of insulin secretion and inhibition of glucose production by the liver [67]. The depletion of MIF can normalise the blood glucose and lactate response following TNF-α- and lipopolysaccharide-induced stress in mice [51].
MIF and type 2 diabetes
Several lines of clinical evidence support a relationship between MIF and type 2 diabetes. Serum concentrations of MIF are higher in patients with type 2 diabetes than in patients with impaired glucose tolerance (as assessed by an OGTT), who have higher levels than healthy individuals [68–70]. In addition, metabolic tests performed on Pima Indians, an ethnic group prone to type 2 diabetes, have demonstrated a link between MIF and insulin resistance [69]. The association of type 2 diabetes with MIF appears to be strong and closer than that with other proteins, such as C-reactive protein and IL-6 [70].
Interestingly, blood concentrations of MIF are not uniformly distributed between sexes. In a recent large case-cohort study, elevated serum levels of MIF were associated with a higher risk of type 2 diabetes in women (HR 1.74) but not in men [71]. Another factor that may explain the variability of MIF expression in the population is the finding that a number of different MIF promoter genotypes have been identified [72, 73]. For example, a common polymorphism containing between five and eight CATT tetranucleotide repeats (−794 CATT5–8) demonstrated a reduced MIF promoter activity [72]. Similarly, the observed difference in serum levels of MIF between male and female patients with type 2 diabetes has been linked to specific single nucleotide polymorphisms (rs2070767, rs1007888) in the MIF gene [71]. Besides these genetic variations, the difference between sexes may reflect the influence of sex hormones on MIF at the transcriptional level. For instance, it has been shown that oestrogens can regulate MIF production from mouse and human monocytes and macrophages through the important transcriptional regulator nuclear factor κB [74]. This sex specificity is in line with similar observations regarding body composition, serum markers of inflammation and the risk of type 2 diabetes [75].
Serum MIF is correlated with BMI [76], with higher serum protein levels and an increased frequency of certain MIF variants observed in obese individuals [71, 77, 78]. This is supported by the observation that adipocytes from obese donors tested ex vivo secrete higher levels of MIF than adipocytes from non-obese donors [50]. Interestingly, treatment of obese individuals with normal blood glucose levels with metformin (1000 mg orally twice daily) resulted in a significant decrease in plasma MIF concentrations [76]. Similarly, obese individuals participating in physical activity and following a dietary-focused management programme also demonstrated decreased plasma levels of MIF [77]. While obesity is associated with a chronic inflammatory response, MIF is only one of many cytokine mediators, such as TNF-α, IL-1β, IL-6, IL-8, IL-10, TGF-β and nerve growth factor, to be involved in this response [79]. As such, the causal relationship between obesity and MIF production has not been firmly established as yet. In addition, the increased secretion of MIF may originate from various cells and tissues. For example, mononuclear cells of obese individuals demonstrate higher levels of MIF [76].
These clinical studies demonstrate a strong relationship between MIF and type 2 diabetes. However, it has yet to be determined whether increased MIF levels lead to disease development or are instead secondary to type 2 diabetes. Interestingly, a preliminary report suggested a causal effect of MIF [80]. MIF KO mice demonstrated an age-dependent significant weight gain, together with elevations in glucose levels and insulin release following an intra-peritoneal glucose challenge. This type 2 diabetes-like pattern may suggest a central role of MIF in the disease, but further investigations are needed before any conclusions can be drawn.
MIF and type 1 diabetes
MIF levels are lower in patients with recent-onset type 1 diabetes who have multiple autoantibodies (anti-GAD, anti-insulinoma-associated antigen-2, anti-islet cell antibodies). While classical Th1/Th2 cytokines (IFN-γ, IL-5, IL-10, IL-13) are not affected, decreased levels of MIF, together with decreased MCP-1 and increased IL-18, can predict autoantibody positivity with 85% sensitivity and 94% specificity [81]. Interestingly, MIF and MCP-1, usually associated with the innate pathway, are also known to be produced by pancreatic islets [44, 82]. As such, the decrease in these islet-produced hormones could reflect a modification in overall immune activity and/or be associated with decreased islet secretion, which is potentially linked to diabetic injuries. It remains unclear how serum MIF levels of patients with type 1 diabetes compare with those in healthy individuals.
Several lines of experimental data suggest a key role for MIF in the development of autoimmune diabetes. The expression of Mif and release of the protein are increased in both pancreatic islets and peripheral lymphocytes of animals with autoimmune diabetes [83, 84]. In addition, in NOD mice treated with recombinant MIF protein (25 μg i.p. twice a week from 6–11 weeks of age) the incidence of diabetes is increased from 55% to 86% [84]. Consistent with this, several animal experiments have demonstrated that anti-MIF antibody and knockout of Mif could prevent autoimmune diabetes in mice [39, 83]. These observations are specific to autoimmune models of diabetes, since no protective effect was observed after chemical induction of diabetes with a standard dose of streptozotocin i.p. in C57BL/6 mice [83]. Taken together, these results from various animal models suggest a central role for MIF in the development of autoimmune diabetes.
Antagonising the action of MIF has a profound effect on various immune parameters. In mouse models of autoimmune diabetes, the production of proinflammatory mediators in splenic mononuclear cells (TNF-α, IL-1β, IL-12, IL-23, IFN-γ and nitric oxide) and islets (TNF-α, IL-18, IL-1β, and iNOS) is decreased [39, 83]. In contrast, the levels of IL-4, TGF-β and IL-10 are increased, demonstrating an overall immune deviation towards a protective type 2 response. Anti-MIF therapy also leads to decreased expression of the IL-2 receptor on the surface of splenocytes and a lower rate of proliferation of lymphocytes in vitro. These observations suggest that MIF contributes to the clonal expansion of T cells during autoimmune diabetes [39, 83]. Resistance to autoimmune diabetes in MIF KO animals may also be related to observed decreases in susceptibility to injury and apoptosis [85]. These animals also demonstrate lower levels of production of IL-1β [85].
Overall, these observations suggest that anti-MIF strategies can prevent autoimmune diabetes in mice. While this represents a very important preliminary step, it should be noted that these animal models only approximate the human disease. The demonstration of the efficiency of a treatment in mice is therefore not a guarantee of success in humans.
Besides type 1 diabetes, MIF may also play a role in other less common variations of the disease. This is the case for MODY3, which is caused by heterozygous mutations in the gene encoding hepatocyte nuclear factor-1α (HNF1A) and is characterised by impaired insulin secretion. INS-1 cells engineered to overexpress HNF1A containing a mutation associated with MODY3 in humans showed impaired growth and decreased expression of Mif [86].
MIF and islet transplantation
The impact of MIF on the allogeneic immune response in transplantation is controversial. MIF production is upregulated in cases of kidney or heart graft rejection, as demonstrated by animal and human studies [87–89]. Neutralising anti-MIF antibodies can inhibit indirect allorecognition of skin grafts, probably partly through inhibition of macrophages [41]. Blockade of MIF production with a liposome-transfected small interfering RNA prevented early obstructive bronchiolitis and destruction in a mouse tracheal allograft model [90]. Conversely, murine cardiac and renal allograft rejections were not prevented or even delayed in MIF KO recipients or by use of anti-MIF neutralising antibodies [42, 89].
The challenges linked to islets differ from those related to solid organ transplantation. First, pancreatic islets produce MIF [44, 45, 91], which can have a local impact at the site of islet implantation. Microarray studies have demonstrated that MIF is one of the most strongly expressed genes in human islets in culture [91]. Furthermore, islet transplantation involves the early loss of many islet cells (50% or more), owing to poor engraftment. Several factors may induce these events, including the activation of the coagulation cascade, which is responsible for non-specific inflammatory reactions [92]. Several mediators, including MCP-1, IFN-γ and IL-1β, have been associated with lower islet isolation yields and impaired post-transplantation islet function [82, 93]. Based on these findings, one can postulate that MIF is involved in a similar deleterious process. This hypothesis is supported by a report of decreased in vitro cytokine-induced death in MIF-depleted islets [85].
Finally, since islet transplantation is most often performed in patients with type 1 diabetes, some patients will develop a progressive recurrence of autoimmunity to the islet graft [94]. Since MIF plays a role in the development of autoimmune diabetes in animal models, it is possible that an intervention aiming at inhibiting MIF secretion may also improve the long-term survival of the islet grafts. Potential strategies include the use anti-MIF antibodies or molecular inhibitors such as the recently developed 3-(4-hydroxyphenyl)-4,5-hydro-5-isoazole acetic acid methylester (ISO-1) [95, 96]. These strategies remain to be tested in the setting of islet transplantation.
Summary and future directions
MIF is an important mediator involved in numerous immuno-inflammatory disorders and pathways. For this reason, many efforts have been made to develop neutralising strategies that can potentially be applied to humans [96]. The use of neutralising anti-MIF antibody has proven to be efficient in several animal models, including septic shock, arthritis, multiple sclerosis (experimental autoimmune encephalomyelitis) and asthma [96]. Chemical inhibitors such as ISO-1 or COR100140 have also been developed and tested in animal models of inflammatory disorders [96]. The modes of action of these compounds still remain to be fully elucidated and their side effect profiles better characterised, but they may be interesting tools for future interventions on diabetes.
With its dual proinflammatory and metabolic effects, MIF represents a logical focus of study in the field of diabetes. An association between type 2 diabetes and MIF appears clear, with higher plasma MIF levels observed in affected individuals than in healthy controls [70, 71]. While type 2 diabetes is associated with obesity, and is itself related to higher circulating levels of many proinflammatory cytokines, the main challenge will be to determine whether a causal relationship exists between MIF and type 2 diabetes. If this were proved to be the case, a new therapeutic target could be developed. In murine models of type 1 diabetes, the inhibition of MIF modulated the immune response involved in the disease and maintained stable blood glucose levels [39, 83]. These experiments demonstrate a direct causal role of the cytokine in this autoimmune disease. While important, the mechanisms associated with these observations remain to be fully characterised.
Although MIF appears to be a potential therapeutic candidate for these diseases, one must keep in mind the various, sometimes diverging metabolic effects of this cytokine. Besides modulating the immune response and preventing the inflammatory response, inhibition of MIF has the potential to improve the glucose profile by decreasing peripheral insulin resistance [51, 53]. Conversely, MIF inhibition could decrease insulin release from pancreatic beta cells and glucose uptake and glycolysis in muscle and heart.
Overall, the balance between positive therapeutic effects and potential side effects of a MIF-targeting intervention need to be better understood. However, recently, an increasing number of studies have focused on MIF, which has emerged as an important player in the physiological and pathological regulation of glucose metabolism. Taken together, the data reported to date are promising and should encourage further exploration in this field.
Abbreviations
- ACTH:
-
adrenocorticotropic hormone
- ISO-1:
-
3-(4-hydroxyphenyl)-4,5-hydro-5-isoazole acetic acid methylester
- MCP-1:
-
monocyte chemotaxis protein-1
- MIF:
-
macrophage migration inhibitory factor
- PFK-2:
-
6-phosphofructo-2-kinase
- Th1:
-
T helper cell type 1
References
Bloom BR, Bennett B (1966) Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 153:80–82
David JR (1966) Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell–antigen interaction. Proc Natl Acad Sci USA 56:72–77
Lind M, Trindade MC, Nakashima Y, Schurman DJ, Goodman SB, Smith L (1999) Chemotaxis and activation of particle-challenged human monocytes in response to monocyte migration inhibitory factor and C–C chemokines. J Biomed Mater Res 48:246–250
Weiser WY, Temple PA, Witek-Giannotti JS, Remold HG, Clark SC, David JR (1989) Molecular cloning of a cDNA encoding a human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A 86:7522–7526
Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R (1994) Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry 33:14144–14155
Mitchell R, Bacher M, Bernhagen J, Pushkarskaya T, Seldin MF, Bucala R (1995) Cloning and characterization of the gene for mouse macrophage migration inhibitory factor (MIF). J Immunol 154:3863–3870
Budarf M, McDonald T, Sellinger B, Kozak C, Graham C, Wistow G (1997) Localization of the human gene for macrophage migration inhibitory factor (MIF) to chromosome 22q11.2. Genomics 39:235–236
Nathan CF, Karnovsky ML, David JR (1971) Alterations of macrophage functions by mediators from lymphocytes. J Exp Med 133:1356–1376
Nathan CF, Remold HG, David JR (1973) Characterization of a lymphocyte factor which alters macrophage functions. J Exp Med 137:275–290
Calandra T, Bernhagen J, Mitchell RA, Bucala R (1994) The macrophage is an important and previously unrecognized source of macrophage migration inhibitor factor. J Exp Med 179:1895–1902
Meinhardt A, Bacher M, McFarlane JR et al (1996) Macrophage migration inhibitory factor production by Leydig cells: evidence for a role in the regulation of testicular function. Endocrinology 137:5090–5095
Murakami H, Akbar SM, Matsui H, Horiike N, Onji M (2002) Macrophage migration inhibitory factor activates antigen-presenting dendritic cells and induces inflammatory cytokines in ulcerative colitis. Clin Exp Immunol 128:504–510
Daryadel A, Grifone RF, Simon HU, Yousefi S (2006) Apoptotic neutrophils release macrophage migration inhibitory factor upon stimulation with tumor necrosis factor-alpha. J Biol Chem 281:27653–27661
Calandra T, Roger T (2003) Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev 3:791–800
Waeber G, Calandra T, Bonny C, Bucala R (1999) A role for the endocrine and pro-inflammatory mediator MIF in the control of insulin secretion during stress. Diabetes Metab Res Rev 15:47–54
Petrovsky N, Socha L, Silva D, Grossman AB, Metz C, Bucala R (2003) Macrophage migration inhibitory factor exhibits a pronounced circadian rhythm relevant to its role as a glucocorticoid counter-regulator. Immunol Cell Biol 81:137–143
Sun HW, Bernhagen J, Bucala R, Lolis E (1996) Crystal structure at 2.6-Å resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci USA 93:5191–5196
Mitchell RA, Metz CN, Peng T, Bucala R (1999) Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem 274:18100–18106
Kleemann R, Hausser A, Geiger G et al (2000) Intracellular action of the cytokine MIF to modulate AP-1 activity and the cell cycle through Jab1. Nature 408:211–216
Leng L, Metz CN, Fang Y et al (2003) MIF signal transduction initiated by binding to CD74. J Exp Med 197:1467–1476
Fingerle-Rowson G, Petrenko O, Metz CN et al (2003) The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci U S A 100:9354–9359
Shi X, Leng L, Wang T et al (2006) CD44 is the signaling component of the macrophage migration inhibitory factor–CD74 receptor complex. Immunity 25:595–606
Ponta H, Sherman L, Herrlich PA (2003) CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol 4:33–45
Wu J, Cunha FQ, Liew FY, Weiser WY (1993) IL-10 inhibits the synthesis of migration inhibitory factor and migration inhibitory factor-mediated macrophage activation. J Immunol 151:4325–4332
Mitchell RA, Liao H, Chesney J et al (2002) Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci USA 99:345–250
Roger T, David J, Glauser MP, Calandra T (2001) MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 414:920–924
Roger T, Froidevaux C, Martin C, Calandra T (2003) Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4). J Endotoxin Res 9:119–123
Toh ML, Aeberli D, Lacey D et al (2006) Regulation of IL-1 and TNF receptor expression and function by endogenous macrophage migration inhibitory factor. J Immunol 177:4818–4825
Amin MA, Haas CS, Zhu K et al (2006) Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFκB. Blood 107:2252–2261
Bernhagen J, Krohn R, Lue H et al (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med 13:587–596
Gregory JL, Morand EF, McKeown SJ et al (2006) Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol 177:8072–8079
Aeberli D, Yang Y, Mansell A, Santos L, Leech M, Morand EF (2006) Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett 580:974–981
Bacher M, Metz CN, Calandra T et al (1996) An essential regulatory role for macrophage migration inhibitory factor in the T-cell activation. Proc Natl Acad Sci USA 93:7849–7854
Abe R, Peng T, Sailors J, Bucala R, Metz CN (2001) Regulation of the CTL response by macrophage migration inhibitory factor. J Immunol 166:747–753
Yan X, Orentas RJ, Johnson BD (2006) Tumor-derived macrophage migration inhibitory factor (MIF) inhibits T lymphocyte activation. Cytokine 33:188–198
Gore Y, Starlets D, Maharshak N et al (2008) Macrophage migration inhibitory factor induces B cell survival by activation of a CD74–CD44 receptor complex. J Biol Chem 283:2784–2792
Rodriguez-Sosa M, Rosas LE, David JR, Bojalil R, Satoskar AR, Terrazas LI (2003) Macrophage migration inhibitory factor plays a critical role in mediating protection against the helminth parasite Taenia crassiceps. Infect Immun 71:1247–1254
Mizue Y, Ghani S, Leng L et al (2005) Role for macrophage migration inhibitory factor in asthma. Proc Natl Acad Sci USA 102:14410–14415
Stosic-Grujicic S, Stojanovic I, Maksimovic-Ivanic D et al (2008) Macrophage migration inhibitory factor (MIF) is necessary for progression of autoimmune diabetes mellitus. J Cell Physiol 215:665–675
Bernhagen J, Bacher M, Calandra T et al (1996) An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J Exp Med 183:277–282
Hou G, Valujskikh A, Bayer J, Stavitsky AB, Metz C, Heeger PS (2001) In vivo blockade of macrophage migration inhibitory factor prevents skin graft destruction after indirect allorecognition. Transplantation 72:1890–7
Jose MD, David JR, Akins C, Chadban SJ (2003) Blockade of macrophage migration inhibitory factor does not prevent acute renal allograft rejection. Am J Transplant 3:1099–1106
Allaman-Pillet N, Roduit R, Oberson A et al (2004) Circadian regulation of islet genes involved in insulin production and secretion. Mol Cell Endocrinol 226:59–66
Waeber G, Calandra T, Roduit R et al (1997) Insulin secretion is regulated by the glucose-dependent production of islet beta cell macrophage migration inhibitory factor. Proc Natl Acad Sci U S A 94:4782–4787
Plaisance V, Thompson N, Niederhauser G et al (2002) The mif gene is transcriptionally regulated by glucose in insulin-secreting cells. Biochem Biophys Res Commun 295:174–181
Hirokawa J, Sakaue S, Tagami S et al (1997) Identification of macrophage migration inhibitory factor in adipose tissue and its induction by tumor necrosis factor-alpha. Biochem Biophys Res Commun 235:94–98
Hirokawa J, Sakaue S, Furuya Y et al (1998) Tumor necrosis factor-alpha regulates the gene expression of macrophage migration inhibitory factor through tyrosine kinase-dependent pathway in 3T3-L1 adipocytes. J Biochem 123:733–739
Sakaue S, Nishihira J, Hirokawa J et al (1999) Regulation of macrophage migration inhibitory factor (MIF) expression by glucose and insulin in adipocytes in vitro. Mol Med 5:361–371
Benigni F, Atsumi T, Calandra T et al (2000) The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest 106:1291–1300
Skurk T, Herder C, Kraft I, Muller-Scholze S, Hauner H, Kolb H (2005) Production and release of macrophage migration inhibitory factor from human adipocytes. Endocrinology 146:1006–1011
Atsumi T, Cho YR, Leng L et al (2007) The proinflammatory cytokine macrophage migration inhibitory factor regulates glucose metabolism during systemic inflammation. J Immunol 179:5399–5406
Miller EJ, Li J, Leng L et al (2008) Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 451:578–582
Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM (1994) Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A 91:4854–4858
Larson DF, Horak K (2006) Macrophage migration inhibitory factor: controller of systemic inflammation. Crit Care 10:138
Gando S, Nishihira J, Kobayashi S, Morimoto Y, Matsushita M, Kemmotsu O (2001) Systemic macrophage migration inhibitory factor release following hepatic resection. Surg Today 31:605–609
Gando S, Nishihira J, Kobayashi S, Morimoto Y, Nanzaki S, Kemmotsu O (2001) Macrophage migration inhibitory factor is a critical mediator of systemic inflammatory response syndrome. Intensive Care Med 27:1187–1193
Emonts M, Sweep FC, Grebenchtchikov N et al (2007) Association between high levels of blood macrophage migration inhibitory factor, inappropriate adrenal response, and early death in patients with severe sepsis. Clin Infect Dis 44:1321–1328
Flaster H, Bernhagen J, Calandra T, Bucala R (2007) The macrophage migration inhibitory factor-glucocorticoid dyad: regulation of inflammation and immunity. Mol Endocrinol 21:1267–1280
Bacher M, Meinhardt A, Lan HY et al (1997) Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol 150:235–246
Nishino T, Bernhagen J, Shiiki H, Calandra T, Dohi K, Bucala R (1995) Localization of macrophage migration inhibitory factor (MIF) to secretory granules within the corticotrophic and thyrotrophic cells of the pituitary gland. Mol Med 1:781–788
Tampanaru-Sarmesiu A, Stefaneanu L, Thapar K et al (1997) Immunocytochemical localization of macrophage migration inhibitory factor in human hypophysis and pituitary adenomas. Arch Pathol Lab Med 121:404–410
Waeber G, Thompson N, Chautard T et al (1998) Transcriptional activation of the macrophage migration-inhibitory factor gene by the corticotropin-releasing factor is mediated by the cyclic adenosine 3′,5′-monophosphate responsive element-binding protein CREB in pituitary cells. Mol Endocrinol 12:698–705
Fingerle-Rowson G, Koch P, Bikoff R et al (2003) Regulation of macrophage migration inhibitory factor expression by glucocorticoids in vivo. Am J Pathol 162:47–56
Bernhagen J, Calandra T, Mitchell RA et al (1993) MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 365:756–759
Calandra T, Echtenacher B, Le Roy D et al (2000) Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med 6:164–70
Lang CH, Dobrescu C, Meszaros K (1990) Insulin-mediated glucose uptake by individual tissues during sepsis. Metabolism 39:1096–1107
Miller SI, Wallace RJ, Musher DM, Septimus EJ, Kohl S, Baughn RE (1980) Hypoglycemia as a manifestation of sepsis. Am J Med 68:649–654
Yabunaka N, Nishihira J, Mizue Y et al (2000) Elevated serum content of macrophage migration inhibitory factor in patients with type 2 diabetes. Diabetes Care 23:256–258
Vozarova B, Stefan N, Hanson R et al (2002) Plasma concentrations of macrophage migration inhibitory factor are elevated in Pima Indians compared to Caucasians and are associated with insulin resistance. Diabetologia 45:1739–1741
Herder C, Kolb H, Koenig W et al (2006) Association of systemic concentrations of macrophage migration inhibitory factor with impaired glucose tolerance and type 2 diabetes: results from the Cooperative Health Research in the Region of Augsburg, Survey 4 (KORA S4). Diabetes Care 29:368–371
Herder C, Klopp N, Baumert J et al (2008) Effect of macrophage migration inhibitory factor (MIF) gene variants and MIF serum concentrations on the risk of type 2 diabetes: results from the MONICA/KORA Augsburg Case–Cohort Study, 1984–2002. Diabetologia 51:276–284
Baugh JA, Chitnis S, Donnelly SC et al (2002) A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun 3:170–176
Zhong XB, Leng L, Beitin A et al (2005) Simultaneous detection of microsatellite repeats and SNPs in the macrophage migration inhibitory factor (MIF) gene by thin-film biosensor chips and application to rural field studies. Nucleic Acids Res 33:e121
Hardman MJ, Waite A, Zeef L, Burow M, Nakayama T, Ashcroft GS (2005) Macrophage migration inhibitory factor: a central regulator of wound healing. Am J Pathol 167:1561–1574
Thorand B, Baumert J, Kolb H et al (2007) Sex differences in the prediction of type 2 diabetes by inflammatory markers: results from the MONICA/KORA Augsburg Case–Cohort Study, 1984–2002. Diabetes Care 30:854–860
Dandona P, Aljada A, Ghanim H et al (2004) Increased plasma concentration of macrophage migration inhibitory factor (MIF) and MIF mRNA in mononuclear cells in the obese and the suppressive action of metformin. J Clin Endocrinol Metab 89:5043–5047
Church TS, Willis MS, Priest EL et al (2005) Obesity, macrophage migration inhibitory factor, and weight loss. Int J Obes (Lond) 29:675–681
Sakaue S, Ishimaru S, Hizawa N et al (2006) Promoter polymorphism in the macrophage migration inhibitory factor gene is associated with obesity. Int J Obes (Lond) 30:238–242
Trayhurn P, Wood IS (2004) Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 92:347–355
Serre-Beiner V, Toso C, Morel P et al (2007) Macrophage migration inhibitory factor (MIF) plays an important role in the maintenance of glucose homeostasis. Diabetologia 50:S27
Hanifi-Moghaddam P, Schloot NC, Kappler S, Seissler J, Kolb H (2003) An association of autoantibody status and serum cytokine levels in type 1 diabetes. Diabetes 52:1137–1142
Piemonti L, Leone BE, Nano R et al (2002) Human pancreatic islets produce and secrete MCP-1/CCL2: relevance in human islet transplantation. Diabetes 51:55–65
Cvetkovic I, Al-Abed Y, Miljkovic D et al (2005) Critical role of macrophage migration inhibitory factor activity in experimental autoimmune diabetes. Endocrinology 146:2942–2951
Bojunga J, Kusterer K, Bacher M, Kurek R, Usadel KH, Renneberg H (2003) Macrophage migration inhibitory factor and development of type-1 diabetes in non-obese diabetic mice. Cytokine 21:179–186
Stojanovic I, Lazaroski S, Nicoletti F, Stosic-Grujicic S (2007) Lack of macrophage migration inhibitory factor (MIF) confers resistance to immunoinflammatory diabetes in mice. Diabetologia 50:S187
Yang Q, Yamagata K, Fukui K et al (2002) Hepatocyte nuclear factor-1alpha modulates pancreatic beta-cell growth by regulating the expression of insulin-like growth factor-1 in INS-1 cells. Diabetes 51:1785–1792
Brown FG, Nikolic-Paterson DJ, Metz C, Bucala R, Atkins RC, Lan HY (1999) Up-regulation of macrophage migration inhibitory factor in acute renal allograft rejection in the rat. Clin Exp Immunol 118:329–336
Lan HY, Yang N, Brown FG et al (1998) Macrophage migration inhibitory factor expression in human renal allograft rejection. Transplantation 66:1465–71
Demir Y, Chen Y, Metz C, Renz H, Heeger PS (2003) Cardiac allograft rejection in the absence of macrophage migration inhibitory factor. Transplantation 76:244–247
Fukuyama S, Yoshino I, Yamaguchi M et al (2005) Blockage of the macrophage migration inhibitory factor expression by the short interference RNA inhibited the rejection of an allogeneic tracheal graft. Transplant Int 18:1203–1209
Johansson U, Olsson A, Gabrielsson S, Nilsson B, Korsgren O (2003) Inflammatory mediators expressed in human islets of Langerhans: implications for islet transplantation. Biochem Biophys Res Commun 308:474–479
Johansson H, Lukinius A, Moberg L et al (2005) Tissue factor produced by the endocrine cells of the islets of Langerhans is associated with a negative outcome of clinical islet transplantation. Diabetes 54:1755–1762
Barshes NR, Wyllie S, Goss JA (2005) Inflammation-mediated dysfunction and apoptosis in pancreatic islet transplantation: implications for intrahepatic grafts. J Leukoc Biol 77:587–597
Worcester Human Islet Transplantation Group (2006) Autoimmunity after islet-cell allotransplantation. N Engl J Med 355:1397–1399
Dios A, Mitchell RA, Aljabari B et al (2002) Inhibition of MIF bioactivity by rational design of pharmacological inhibitors of MIF tautomerase activity. J Med Chem 45:2410–2416
Cvetkovic I, Stosic-Grujicic S (2006) Neutralization of macrophage migration inhibitory factor—novel approach for the treatment of immunoinflammatory disorders. Int Immunopharmacol 6:1527–1534
Bozza M, Satoskar AR, Lin G et al (1999) Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med 189:341–346
Calandra T, Bernhagen J, Metz CN et al (1995) MIF as a glucocorticoid-induced modulator of cytokine production. Nature 377:68–71
Acknowledgements
The authors thank D. Colwell for drawing the figures. C. Toso is supported by the Swiss National Science Foundation, the F. S. Chia award and the Alberta Heritage Foundation for Medical Research (AHFMR). J. A. Emamaullee is supported by fellowship awards from the AHFMR, the American Society for Transplantation, the Juvenile Diabetes Research Foundation (JDRF), and the Rhind Foundation. S. Merani is a recipient of the AHFMR MD/PhD Studentship, the Canadian Institutes of Health Research Walter and Jessie Boyd and Charles Scriver MD/PhD Studentship, and the Lionel E. McLeod Award. A. M. J. Shapiro is a scholar supported through the AHFMR, through a Centre Grant from the Juvenile Diabetes Research Foundation and through the Collaborative Islet Transplant Consortium of the National Institutes of Health.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Toso, C., Emamaullee, J.A., Merani, S. et al. The role of macrophage migration inhibitory factor on glucose metabolism and diabetes. Diabetologia 51, 1937–1946 (2008). https://doi.org/10.1007/s00125-008-1063-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1063-3