
Abstract
Human influenza commonly known as seasonal flu which is caused by a RNA virus has been emerging as a major viral infection over the years. Virus neuraminidase inhibitors and M2 protein inhibitors are the agents which have been used to treat this viral infection. Among these two, viral neuraminidases named oseltamivir and zanamivir are most widely used as antiviral agents to treat influenza. But the recent emergence of resistance strains in the treatment with both zanamivir and oseltamivir creates a big problem to treat this viral infection effectively. In this study, we have designed 68 new human influenza virus neuraminidase inhibitors and reported them as new potential antiviral agents against the complex structure of influenza virus neuraminidase and sialic acid using various in silico tools and molecular docking analysis taking zanamivir as prototype.
Similar content being viewed by others

Introduction
Human influenza virus is highly infectious and causes seasonal influenza with the global impact of 3.5 million cases of severe illness and 300,000–500,000 deaths (Fiore et al., 2008). The main enzyme responsible for flu is viral neuraminidase that is found to exist on the surface of influenza viruses and is essential for its replication. For the virus to be released from the cell, neuraminidase must enzymatically cleave the sialic acid groups from host glycoproteins and thus help in detaching the budding viruses. Neuraminidase inhibitors block the action of viral neuraminidase so the viruses will not able to release itself from cell thus stopping viral effect. Some major antiviral agents act against influenza and inhibit the enzyme neuraminidase (Huang et al., 2008). Two major neuraminidase inhibitors commonly used for combating influenza infection are zanamivir and oseltamivir. The discovery of the first designed influenza virus neuraminidase inhibitor and anti-influenza drug zanamivir and subsequently oseltamivir has now inspired a number of continuing efforts toward the discovery of next generation anti-influenza drugs. Such drugs may act as “first-line-of-defense” against the spread of influenza infection and buy time for necessary vaccine development particularly in a human pandemic setting (Itzstein and Thomson, 2009).
But recent report of emergence of oseltamivir and zanamivir resistant viruses creates a great concern in antiviral (influenza) research and offers a challenge to drug designers to design some new neuraminidase inhibitors to be more potent against zanamivir, as zanamivir resistance is quite limited and is the drug of choice in case of oseltamivir resistance (Hurt et al., 2009). Zanamivir and oseltamivir have been designed earlier using the concept of rational drug design and computer-aided drug design tools, but due to recent emergence of resistant strains against both of these, we have designed some new ligands by modifying the structure of zanamivir and oseltamivir and judging the effectiveness against viral neuraminidase using computer-aided drug design approach (Virupakshaiah et al., 2007; Singh et al., 1995). Current computer-aided molecular docking approach has been used in this study to judge the binding efficiency of the new ligands with the neuraminidase macromolecule and comparison of the binding efficiency of new ligands with that of zanamivir, as it is still the drug of choice over oseltamivir, as oseltamivir resistance is quite greater than zanamivir (Bauer et al., 2009; Stephenson et al., 2008).
In this study, we designed some new neuraminidase inhibitors by structure–activity relationship modification which has been proved to possess better molecular property and higher efficiency against neuraminidase receptor than zanamivir.
Materials and methods
Designing of ligands
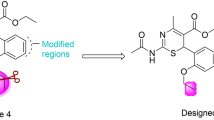
Sixty-two ligands have been designed as potential antiviral (influenza) agents (structure of all the ligands with their codes is attached as supplementary materials). All the ligands were designed by random modification of the basic structure (Fig. 1), which is having structural similarity with either zanamivir or oseltamivir.

Basic skeleton that is used for structure modification
The ligands were designed by modification at positions R1, R2, R3, and R4 and by doing nuclear modification at ring A. The activities of the designed ligands have been predicted using PASS online server (http://www.pharmaexpert.ru/passonline/predict.php) and were used for the prediction of substance activity spectrum (Goel et al., 2011). All molecules were individually subjected to the PASS server and it has been observed that the molecules showed greater probability of active (Pa) than probability of inactive (Pi) as antiviral (Influenza) and neuraminidase inhibitors.
ADME and toxicity prediction
The ADME/T properties of a drug together with its pharmacological properties are conventionally viewed as part of drug development. The best ligands after docking analysis were subjected to pre-ADMET online server (Lee et al., 2003) to predict the pharmacokinetic properties. Structures with unfavorable absorption, distribution, metabolism, and elimination were identified as the major cause of failure of candidate molecules in drug development. So there is an early prediction of ADME properties, with the objective of increasing the success rate of compounds reaching further stages of the development.
Molecular property prediction
As molecular properties are essential for every stages of drug development from design to synthesis, molecular property, Lipinski rule of 5 violation, and other parameters of the best ligand after docking study were predicted by MedChem Designer (Kotsampasakou and Demopoulos, 2013) and Molsoft Molecular Property Prediction web server (http://www.molsoft.com/mprop/).
Molecular docking analysis
All the designed molecules were docked against the human influenza virus-neuraminidase protein (PDB ID: 2BAT) using Molegro virtual docker (MVD) (Thomsen and Christensen, 2006; Varghese et al., 1992). The Molegro scoring system is utilized by MVD and this scoring system is based on a novel hybrid search algorithm, called guided differential evolution, which is a combination of the differential evolution optimization technique together with a cavity prediction algorithm. Rapid and precise identification of potential binding poses is facilitated by use of predicted cavities during the search process. Moldock score scoring function was used. The intact protein structure was loaded on to MVD platform for docking process. Potential binding cavities were identified by selecting interested binding site residue and toward that residue 16 Å constrain was generated. The search algorithm was taken as Moldock SE and number of runs was taken as 10 and max iterations were set to 2,000 with population size 50 and an energy threshold of 100. At each step least “min” torsions/translations/rotations were tested and the one yielding the least energy was taken. After the docking simulation got over, the poses which were generated were sorted by Moldock score. The manual preparation of chosen ligands was carried out with the ligand preparation module of MVD. Wherever bond order and hybridization were missing, they were assigned flexible torsion and the ligands were deducted. The target protein structure—2BAT was prepared after removal of water molecules and hetero atoms and the electrostatic surface of the protein was generated.
Results and discussion
Activity prediction
The Pa and Pi values predicted using PASS server of the best three ligands which were chosen from docking analysis were compared with zanamivir (Table 1).
ADME and toxicity results
The ADME and toxicity parameters of the best three compounds chosen from the docking analysis (SF1, SF2, and SF53) were predicted using preADMET server and compared with the prototype molecule zanamivir and are shown in Tables 2 and 3.
It has been found that SF1 who’s predicted ADME parameters closely resembles zanamivir has poor percentage of intestinal absorption and also bounds weakly to plasma protein, which lead us to the prediction that it can be given through intravenous route for quick onset of action (Jusko and Gretch, 1976). The plasma protein binding of zanamivir is predicted to be less than 90 %, which resembles the fact that practically the plasma protein binding of zanamivir is <10 % proving that the prediction was quite satisfactory and as SF1 resembles with this prediction, demonstrating close matches with zanamivir in terms of plasma protein binding. SF2 also predicted to be poorly absorbed in intestine and its plasma protein binding is also very weak which makes it a better drug candidate when administered through the intravenous route. As ADME parameters are concerned, both SF1 and SF2 have similarly predicted ADME parameters as that of zanamivir. Whereas SF53 had moderate percentage of intestinal absorption and also predicts to be weakly bound with the plasma protein, this makes it a good candidate for oral administration. Also all the molecules except SF1 and SF53 are predicted to have similar toxicity parameters that of the prototype molecule zanamivir but in case of SF1 and SF53 it has been predicted to be non-mutagenic in Ames Test (Mortelmans and Zeiger, 2000) and carcinogenic (in rat) which makes them better and promising candidate as novel neuraminidase inhibitor.
Docking result
In the case of influenza, all drugs were developed based on the knowledge of enzyme structure. It is less likely to select drug-resistant viruses to retain viability if inhibitor has closer structural resemblance to natural substrate (Collins et al., 2008). Oseltamivir (tamiflu) and zanamivir (relenza) are two drugs which are used currently as neuaramidase inhibitor, from which binding of oseltamivir is not that compact, so it results in easy displacement of polysaccharide substrates which make the drug least effective against the mutant virus (Goodsell, 2009). Our aim of the study was to design inhibitor and dock against 2BAT for targeting the highly conserved active site among all the NA subtypes, including eight charged polar residues (Arg118, Asp151, Arg152, Arg224, Glu276, Arg292, Arg371, and Tyr406) which have direct interaction with the substrate at the catalytic site (Xu and Zhu, 2008). Our study with taking zanamivir as prototype showed promising result. For each compound, out of the many docking poses, only those which possessed the highest moldock score and relatively good hydrogen bond interaction against the targeting binding site were chosen. The best three compounds which exhibited a very good affinity, even better than zanamivir, were SF1, SF2, and SF53. From this SF1 binds to the receptor by targeting the residue (Arg118, Glu119, Arg152, Ser179, Ile222, Arg224, Glu227, Ala246, Glu276, Arg 292, Asn294, Gly348, Arg371, Tyr406) with 16 H-bond and with -131.832 moldock score. In the same way, SF2 and SF53 bind to the 2BAT receptor with the 10 and 11 H-bond and with −116.595 and −115.227 moldock score, respectively, to the same targeted site which is better than zanamivir. Results of the same are depicted in Fig. 2 and detailed docking results are listed in Table 4. It has been predicted from the molecular docking approach and ADME/T parameters that SF1, SF2, and SF53 can be considered as potential new candidates targeting viral neuraminidase in a more efficient manner than that of zanamivir with SF1 predicted to be orally active and more efficient than zanamivir, and SF2 and SF53 can be delivered as the same way that of zanamivir but possibly more effective toward target. The proposed theoretical synthetic scheme for preparation of SF53, SF2, and SF1 has been depicted in Figs. 3, 4, and 5, respectively, which we believe will be useful for future development of these molecules.

Illustrations of the docking pose of screened inhibitor binding with 2BAT carried out with Molegro virtual docker 5.0. A1, B1, C1, and D1 show the electrostatic interaction of ligand with the 2BAT receptor in which ligand represented in stick model. Where A2, B2, C2, and D2 show the Hbond interaction with 2BAT where green dotted lines represent hydrogen bonds. Receptor is depicted in ball and stick model and the ligand in wireframe. CPK coloring convention has been employed. A1 and A2—SF1; B1 and B2—SF2; C1 and C3—SF53; D1 and D2—Zanamivir (Color figure online)

Proposed synthetic scheme of SF53

Proposed synthetic scheme of SF2 from zanamivir

Proposed synthetic scheme of SF1 from zanamivir
Conclusion
Our approach was to design the molecule which is similar to the natural substrates (sialic acid) of the enzyme and which binds with more competence to the binding site similar to zanamivir. Our study ended by giving the three molecules which established better result in in silico analysis with better binding efficiency toward viral neuraminidase than that of zanamivir and it would possibly give better results in further justification process which can promisingly lead to discovery of a better neuraminidase inhibitor as a potential new anti-influenza agent.
References
Bauer K, Richter M, Wutzler P, Schmidtke M (2009) Different neuraminidase inhibitor susceptibilities of human H1N1, H1N2, and H3N2 influenza A viruses isolated in Germany from 2001 to 2005/06. Antivir Res 82(1):34–41
Collins PJ, Haire LF, Lin YP, Liu J (2008) Crystal structures of oseltamivir-resistant influenza virus neuaramidase mutants. Nature 453(7199):1258–1261
Fiore AE, Shay DK, Broder K, Iskander JK, Uyeki TM, Mootrey G, Bresee JS, Cox NS, Centers for Disease Control and Prevention (CDC), Advisory Committee on Immunization Practices (ACIP) (2008) Prevention and control of influenza: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2008. MMWR Recomm Rep 57(RR-7):1–60
Goel RK, Singh D, Lagunin A, Poroikov V (2011) PASS-assisted exploration of new therapeutic potential of natural products. Med Chem Res 20(9):1509–1514
Goodsell D (2009) Influenza neuraminidase. doi: 10.2210/rcsb_pdb/mom_2009_5
Huang IC, Li W, Sui J, Marasco W, Choe H, Farzan M (2008) Influenza A virus neuraminidase limits viral super infection. J Virol 82(10):4834–4843
Hurt AC, Holien JK, Parker M, Kelso A, Barr IG (2009) Zanamivir-resistant influenza viruses with a novel neuraminidase mutation. J Virol 83(20):10366–10373
Itzstein MV, Thomson R (2009) Anti-influenza drugs: the development of sialidase inhibitors. Handb Exp Pharmacol 189:111–154
Jusko WJ, Gretch M (1976) Plasma and tissue protein binding of drugs in pharmacokinetics. Drug Metab Rev 5(1):43–140
Kotsampasakou E, Demopoulos VJ (2013) Structure-activity-selectivity relations on the keto-pyrrolyl-difluorophenol aldose reductase inhibitory scaffold. Pharmakeftiki 25(I):24–33
Lee SK, Lee IH, Kim HJ, Chang GS, Chung JE, No KT (2003) The PreADME approach: web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties, EuroQSAR 2002 designing drugs and crop protectants: processes, problems and solutions. Blackwell Publishing, MA, pp 418–420. http://preadmet.bmdrc.org/?option=com_content&view=article&id=44&Itemid=56
Mortelmans K, Zeiger E (2000) The Ames Salmonella/microsome mutagenicity assay. Mutat Res 455(1–2):29–60
Singh S, Jedrzejas MJ, Air GM, Luo M, Laver WG, Brouillette WJ (1995) Structure-based inhibitors of influenza virus sialidase. A benzoic acid leads with novel interaction. J Med Chem 38(17):3217–3225
Stephenson I, Clark TW, Pareek M (2008) Antiviral treatment and prevention of seasonal influenza: a comparative review of recommendations in the European Union. J Clin Virol 42(3):244–248
Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49(11):3315–3321
Varghese JN, McKimm-Breschkin JL, Caldwell JB, Kortt AA, Colman PM (1992) The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins 14:327–332
Virupakshaiah DBM, Kelmani C, Patil R, Hegade P (2007) Computer aided docking studies on antiviral drugs against SARS. PWASET 24:297–299
Xu X, Zhu X (2008) Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J Virol 82(21):10493–10501
Acknowledgments
We would like to extend our sincere thanks to Molegro ApS for giving us a fully functional trial version for a period of 30 days through which all the in silico docking work was carried out.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Bhakat, S., Shaikh, F., Yadav, S. et al. Identification of neuraminidase inhibitors by structure-based screening: promising new leads for influenza. Med Chem Res 23, 2803–2809 (2014). https://doi.org/10.1007/s00044-013-0862-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0862-3