
Abstract
Autism spectrum disorder (ASD) is a heterogeneous clinical condition whose prevalence has grown considerably during the last decade. Genetic factors are thought to underpin the disorder, but common genetic variants and epigenetic mechanisms have been increasingly called into question for the majority of ASD cases. Growing prenatal exposure to new environmental toxicants has been shown to potentially affect brain development, leading to altered cognitive, social, attentive, behavioral, and motor performance. Both epidemiological evidence and mechanistic studies assessing oxidative stress, neuroinflammation, epigenetic alterations, and impaired signal transduction, all observed following neurotoxicant exposure, indeed lend biological plausibility to Gene x Environment interactions, whereby environmental toxicants interacting additively or synergistically with genetic liability, can push prenatal neurodevelopmental processes over the threshold for postnatal ASD expression. Research on environmental contributions to ASD and on specific Gene x Environment interaction models ultimately aims at defining targeted preventive strategies.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) is an extended diagnostic category, which includes individuals with impaired social interaction and communication, as well as repetitive stereotyped behaviors, insistence on sameness and sensory abnormalities [1]. Severity ranges from “low-functioning” cases with absence of spoken language and severe intellectual disability, to “high-functioning” individuals with normal to high Intellective Quotient (IQ), subtle social deficits, and some restricted and obsessive interests. Individuals who display few signs of autism without meeting the full diagnostic criteria belong to the “broad autistic phenotype”, making ASD the categorical extreme of a quantitative continuum of traits present in the general population [2].
During the last decades, ASD prevalence estimates have risen to as much as 113/10,000 children in the USA [3], and 62/10,000 globally [4], corresponding to 1:88 and 1:161 children, respectively. This increase is so prominent that it appears hardly accountable solely to enhanced awareness, greater service availability, and broader diagnostic categories. While genetic components are considered extremely relevant to ASD etiology, candidate genes and copy-number variants currently explain about 20 % of syndromic and non-syndromic ASD cases [5]. This percentage will indeed rise once whole-genome sequencing becomes routinely implemented in the clinic, but it will almost certainly never explain all ASD cases. In fact, common genetic variants seemingly account for at least 50 % of ASD liability, leaving ample room for environmental contributions due to their low penetrance and additive effects [6••].
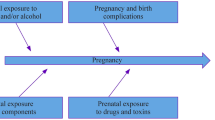
Prenatal exposure to neurotoxic substances has been proven to alter brain maturation and to produce a wide array of neurodevelopmental deficits, in a way that has no counterpart in the adult brain [7]. The developing brain is particularly vulnerable during its “critical periods”, time windows of susceptibility when neuronal proliferation, migration, differentiation, maturation (i.e., neurite sprouting and pruning), synaptogenesis, and activity-dependent synaptic remodelling occur. During these paramount processes, exposure to environmental disruptors can affect brain development, leading to functional deficits and behavioral disorders. In general, a positive epidemiological association, if not spuriously due to mere temporal coincidence, can stem from modulatory, additive, permissive, synergistic, and causal effects exerted by the environmental factor under scrutiny (Fig. 1). Patients not carrying rare, disruptive genetic variants may thus be accounted for by a “multiple-hit” pathogenic model based on complex Gene x Environment interactions, whereby multiple combinations of common genetic variants, each conferring a small risk, create a highly individualized spectrum of sensitivity to the detrimental effects of environmental factors [8]. This conceptual framework can provide a better understanding of the increased prevalence and pathogenetic complexities of ASD, as well as of its amazing clinical heterogeneity.

Environmental factors in human disorders A “none-to-all” upward scaling model, spanning from temporal coincidence without any pathogenic role to full causality. Environmental Factors (EF), in interaction among themselves and with Genetic Factors (GF), can exert: (a) modulatory effects: EF influence the phenotypic expression of GF only qualitatively; (b) permissive effects: one EF is necessary for another EF to exert damage, or for a GF to become penetrant and to achieve phenotypic expression; (c) additive effects: EF and GF act independently and their combined effect equals the sum of each contributing factor; (d) synergistic effects: EF and GF potentiate each other, so that the combined effect is greater than the predicted sum of each contributing factor
The Tip of the Iceberg: Known Environmental Causes of ASD
Case reports, patient cohort studies, epidemiological and neuroanatomical investigations, as well as animal models, have proven that, in a limited number of cases, environmental factors can be regarded as the sole causative agent in ASD. This is the case for some teratogenic drugs, namely valproic acid, misoprostol and thalidomide, as well as for prenatal rubella and cytomegalovirus infections (Table 1).
Teratogenic Drugs
Prenatal exposure to antiepileptic drugs (AED) has been linked to “fetal anticonvulsant syndrome” (FAS), characterised by congenital malformations and developmental delay. Autistic symptoms can also be part of FAS, most frequently following prenatal exposure to sodium valproate (VPA), although some ASD cases have been reported after phenytoin, carbamazepine, or polydrug therapy. Several cohort studies have looked at neurodevelopmental outcomes following prenatal AED exposure, and found increased incidence of ASD, ADHD and dyspraxia, as well as decreased adaptive skills and emotional control, with VPA being most frequently involved [9–11].
VPA teratogenicity in ASD has received unequivocal support by epidemiologic and experimental studies, to the point that mice prenatally exposed to VPA are frequently used as a rodent model of autism. Children prenatally exposed to VPA display hazard ratios of 2.9 % and 5.2 % for ASD and childhood autism, respectively, as defined by the ICD-10, after adjusting for parental psychiatric disease and epilepsy [12]. These rates are significantly higher than the 0.6–1.1 % encountered in the general population [3, 4]. Clinical characteristics of VPA-related autism include an M:F ratio close to 1, minor and major malformations (glue ear, joint laxity, hernias, and congenital defects of the neural tube, heart, genitourinary tract, upper airways, eyes, skin and teeth), significant speech delay, and mild motor delay in the absence of severe cognitive impairment or regression (i.e., loss of acquired skills) [13]. VPA teratogenity can be explained by increased oxidative stress, acid folic deficiency, interference with the Ras-ERK and GSK3β intracellular pathways favouring neuronal differentiation over proliferation, and most importantly by inhibition of histone deacetylase, resulting in persistent histone acetylation and cytosine demethylation at promoters controlling the expression of several neurodevelopmentally-relevant genes, such as WNT, FZD-5, GFRA-2, and GATA-3 [14–16]. Animal models of prenatal VPA exposure show interesting neuroanatomical, behavioral and electrophysiological abnormalities, including: (a) enhanced growth and abnormal distribution of serotoninergic terminals in the brainstem, which reflects defective differentiation and migration of serotoninergic neurons [17]; (b) reduced cerebellar volume, presumably due to diminished Purkinje cells number [18]; (c) local hyperconnectivity in the neocortex, as well as diminished numbers of putative synaptic contacts between layer-5 pyramidal neurons [19]; (d) abnormal fear conditioning and amygdala processing [20], and (e) enhanced expression of NMDA receptors determining increased long-term potentiation [21]. Importantly, the role of epigenetic alterations in VPA-related autism fits with the broad gene expression dysregulation observed in multiple data sets in idiopathic autism [22]. In particular, this dysregulation may implicate genes encoding chromatin-related proteins involved in the transcriptional regulation of prenatal brain development [23].
Thalidomide was used as a sedative and to relieve morning sickness in pregnant women until 1961, when it was withdrawn from the market because of its teratogenic effects. Thalidomide causes multiple systemic malformations, abnormal cortical development and neuronal hyperexcitability, primarily by inhibiting angiogenesis, altering gene expression, broadly perturbing morphoregulatory processes and producing DNA oxidative damage [24, 25]. Similarly to VPA, thalidomide also disrupts early serotoninergic neurodevelopment [17]. Misoprostol is a methyl ester derivative of prostaglandin E1, used to treat gastric ulcers and to induce abortion due to its stimulatory effect on uterine contractions. Its teratogenicity has been observed in children born after unsuccessful abortion attempts, who show cranial nerve hypoplasia sometimes associated with a Möbius sequence, limb malformation and, in some cases, autistic-like behaviors [26]. Thalidomide and misoprostol show many similarities: (a) both alter vascularisation and gene expression, particularly in genes concerned with balancing proliferation and apoptosis, cell migration, neuronal differentiation, and synaptogenesis; (b) both can induce autism, specifically after prenatal exposure very early during pregnancy (embryonic days 18–30 and 18–42 post-fertilisation for thalidomide and misoprostol, respectively); and (c) both affect limbs, ears, eyes, cranial nerves, and the central nervous system (CNS), yielding mental retardation, language impairment and autism [27, 28]. Thalidomide and misoprostol clearly resemble the prenatal exposure timing and phenotypic manifestations of VPA-induced fetal anticonvulsant syndrome. This evidence, in conjunction with the observation that children with autism frequently display minor physical anomalies established during early organogenesis, support the increased importance of an early prenatal time window of maximum sensitivity over the specific nature of the teratogenic agent [29].
Congenital Viral Infections
The two infectious agents most widely acknowledged as conferring ASD risk following congenital infection are rubella and cytomegalovirus (CMV) [30]. Children infected prenatally with these viruses show multisystem impairment and CNS abnormalities ranging from macroscopic neocortical malformations subsequent to migration defects (polymicrogyria, pachygyria, heterotopias), to microscopic alterations in neuronal myelination. Initial evidence linking prenatal rubella infection to ASD came from a longitudinal study involving 448 prenatally infected children [31]. Autism rates were as high as 7.4 % and risk seemed particularly increased when the infection occurred within the first 8 weeks of pregnancy. In addition to autism, deafness, eye defects, cardiopathy and mental retardation were also described. “Late” physical and psychiatric manifestations can appear during adolescence or early adulthood, suggesting that environmental influences can result in clinical courses less stable than those seen in the majority of children with idiopathic ASD.
Several case reports link prenatal cytomegalovirus (CMV) infection to ASDs [31–34]. To what extent autism stems from direct viral damage, from the nature and location of CMV cerebral lesions, or from the strong immune response driven by the virus, still remains to be established [35].
The Submerged Iceberg: Environmental Factors Potentially Involved in ASD
The number of potential teratogens is rapidly increasing, and developmental neurotoxicity has already been ascertained for over 200 agents [36]. The developing brain is especially sensitive to neurotoxicants [37], and preliminary evidence links neurodevelopmental disorders to early exposure to several neurotoxic agents [36]. Environmental factors potentially involved in autism pathogenesis are listed in Table 2 and discussed below.
Air Pollution
Air pollution is comprised of a diverse mixture of particulate matter (PM), gases (e.g., ground-level ozone, carbon monoxide, sulphur oxide), organic compounds (e.g., polycyclic aromatic hydrocarbons) and metals (e.g., nickel, manganese) present in outdoor and indoor air; many millions of people around the world are chronically exposed to air pollutants above promulgated safety standards [38]. Emissions from mobile and residential fuels, wood combustion, construction and demolition works, and industrial activities (e.g., refineries, metal processing facilities) are some examples of air pollution sources.
Prenatal exposure to air pollution has been found to increase risk for low birth weight, pre-term birth, and postnatal mortality [39, 40]; later in life, low performances on cognitive, attentive and memory tests have been described [41, 42]. Interestingly, genetically susceptible children appear to suffer greater impairment [42], supporting Gene x Environment interactions with synergistic or additive mechanisms. Large-scale epidemiological investigations have shown increased autism risk in children prenatally exposed to air pollutants [43–46]. In the large cohort of the CHARGE study, a dispersion model of traffic-related pollutants, combined with measurements of nitrogen dioxide (NO2), ozone (O3), and particulate matter (PM10 and PM25) air concentrations, estimated a two-fold increase in ASD risk among children prenatally exposed to traffic-related air pollution and a three-fold increase when exposure occurred also during their first year of postnatal life [45]. Even when considered individually, exposure to PM25, PM10, or NO2 was associated with a two-fold increase in ASD risk [45]. A different cohort of 7,603 children with autism and 10 matched controls per case, all born between 1995 and 2006 to mothers residing in Los Angeles county at the time of child birth, yielded relative increases in the odds of an ASD diagnosis per interquartile range (IQR) of increased exposure to NO/NO2, PM25 and O3 at 3–9 %, 5–15 % and 6–12 %, respectively, confirming previous results but with smaller effect sizes [46]. These air pollutants mostly derive from traffic, and their association with autism is strongest in the offspring of mothers with low educational level and poor socio-economic status [46].
Neurodevelopmental damage following early exposure to air pollution may ensue from several cellular and molecular mechanisms, depending on the chemical and physical characteristics of each pollutant. In general, oxidative stress, microglial activation, neuroinflammation, cerebral vascular damage and neurodegeneration have all been found after air pollution exposure [47, 48]. Inflammatory damage may follow direct exposure to air pollutants, reaching the CNS through either the olfactory mucosa or systemic circulation, or indirectly by the action of proinflammatory cytokines, released in the respiratory system [48]. Damage to the blood–brain barrier can then enhance pollutant access to the CNS and generate further neural damage in a vicious cycle [47, 48].Animal and cellular models have shown that: (a) dopaminergic neurons are particularly sensitive to the effect of acute and chronic exposure to O3 and to diesel exhaust particles (DEP), which activate microglial cells, causing CNS oxidative damage and cell death in the striatum and substantia nigra [49, 50]; (b) behaviorally, prenatal DEP exposure increases locomotor activity, rearing behaviors, and self-grooming in the presence of an unfamiliar mouse, rodent analogues of the restricted and repetitive patterns of behavior seen in human autism [51]; (c) prenatal or early postnatal exposure during lactation to polycyclic aromatic hydrocarbons (PAH) like benzo(a)pyrene alters DNA methylation and the postnatal expression of ASD-relevant genes, like MET and the 5HT1A receptor gene, producing persistent behavioral abnormalities [52••, 53]. Interestingly, altered DNA methylation has been found also in human cord blood after prenatal exposure to benzo(a)pyrene [54].
Collectively, these data suggest that prenatal and early postnatal exposure to air pollutants can trigger oxidative, inflammatory, and epigenetic alterations in the CNS. In turn, these effects enhance autism risk, provided the exposure occurs during neurodevelopment, is sufficiently prolonged, and likely acts upon genetically susceptible feto-maternal units. The major current limitation of the studies to date is the difficulty to disentangle the effect of air pollution from those of other neurotoxicants potentially more present in California among pregnant women of low education and socioeconomic status.
Insecticides and Pesticides
Organophosphates (OPs) are the most widely used pesticides in agriculture, as well as insecticides in residential, commercial and industrial settings, after organochloride banning. Children may be exposed to OPs via the placenta or through breast milk, food, and inhalation, and appear particularly vulnerable to OPs and to oxidative stress compared to adults, because of their lower activity levels of the enzyme paraoxonase, involved in OP inactivation and lipid peroxide degradation [55]. Prenatal exposure to OPs has been linked to early neurodevelopmental deficits, which appear to be maintained during childhood, including deficits in cognitive abilities, working memory and perceptual reasoning [56–58]. Increased cortical surface in brain regions involved in attention, receptive language, social cognition, reward and behavioral inhibition has been correlated with lower IQ scores in humans prenatally exposed to chlorpyrifos [59•]. Increased risk of developing ASD following prenatal OP exposure has also been reported [60].
OPs inhibit the enzyme acetylcholinesterase (AChE), determining excessive cholinergic transmission; however, OPs main neurotoxic actions are seemingly exerted by their oxon metabolites [61]. At toxicologically relevant doses, these compounds disrupt neuronal proliferation, differentiation, gliogenesis and apoptosis by interfering directly with cell signaling molecules, or indirectly with AchE morphogenetic activities, which are distinct from its enzymatic activity [61]. At the molecular level, OPs and oxons impair neuroligin-neurexin-SHANK signalling, decrease the expression of BDNF and other neurotrophins, interfere with Ca2+-dependent signaling and with the PI3K/mTOR pathway, and disrupt GABAergic neurotransmission, ultimately resulting in altered neuronal connectivity [62, 63]. These effects are predicted to be most functionally significant in genetically vulnerable individuals. For example, OPs inhibit Reelin’s proteolytic activity, crucial for neuronal migration, dendritic spine maturation, and synaptic function [64, 65]. This effect, in conjunction with differences in OP clearance due to interindividual variation in paraoxonase (PON1) activity [66, 67], modulate neurodevelopmental abnormalities, albeit in ways more complex than initially suggested [68]. Also, inflammatory imbalances have been observed after prenatal OP exposure, with upregulation of TH1 and TH2 cytokines in the periphery, and increased IL-6 and IL-1β levels in the CNS [69].
Halogenated Aromatic Hydrocarbons
High hydrophobicity and long half-lives are key features of halogenated aromatic hydrocarbons, a large family of chemicals broadly spread in the environment. Until their banning in 1977, polychlorinated biphenyls (PCBs) were used as coolants, lubricants, and in building materials, whereas polybrominated diphenyl ethers (PBDEs) are flame retardants, still largely employed on infant products, electronic items, and furniture. Early exposure may occur through the placenta and breast milk [70, 71], while postnatal exposure may result from hand-to-mouth behaviors, since these compounds are semivolatile, and accumulate on household surfaces and dust. Impairment in cognition, attention, and motor development has been recorded following prenatal exposure to PBDEs and PCBs [72–74]. Moreover PCBs and PBDEs can alter endocrine and immune functions, as observed in animal models and human studies [75–78]. Multiple neuronal populations (e.g., cerebellar granule cells, hippocampal neurons, nigrostriatal dopaminergic cells) are affected by PBDEs neurotoxic proprieties, apparently due to oxidative stress [79, 80]. On the other hand PCBs produce neurotoxic effects by opening ryanodine receptors (RyR), resulting in enhanced and/or prolonged cytosolic Ca2+ spikes [81]. Excessive and/or prolonged cytosolic Ca2+ spikes can also account for the excessive, spontaneous and the blunted, experience-dependent dendritic growth observed in developing neuronal cells of rats prenatally exposed to PCBs [82, 83•]. Moreover, Gene x Environment interactions involving autism-related genes relevant to Ca2+ management, such as ATP2B2 and SLC25A12 [84, 85•, 86], and PCB exposure could result in altered neuronal connectivity [62]. Interestingly, Gene x Environment interactions converging onto epigenetic alterations relevant to ASD have been found in animal models. Mice carrying a truncated form of the MECP2 gene (i.e., Mecp2308 with a premature stop codon after codon 308), which causes Rett syndrome in humans, after prenatal exposure to PBDEs display globally reduced DNA methylation and enhanced deficits in social behaviour and learning abilities [87].
A limited number of human studies have directly addressed the potential role of PCBs and PBDEs in autism, yielding inconclusive results. No difference in PBDE plasma concentrations have been found in children with autism contrasted with typical or delayed development [88]. Similarly, no difference in PCB levels have been recorded in archived serum samples of Finnish mothers pregnant with 75 children that were later diagnosed with ASD compared to 75 matched controls, although the adjusted model yielded a promising O.R. = 1.92 for autism (P = 0.29), which deserves an independent replication and extension [89]. Curiously, higher amounts of PCB95 were found in postmortem brains of children with maternal 15q11-q13 duplication or deletion compared to brains of children with non-syndromic autism or typical development (TD) [90]. These same brains exhibited lower levels of repetitive DNA methylation, suggesting that exposure to PCB95 might contribute to the generation of specific CNVs [90]. Finally, altered immune responses following LPS stimulation have been observed in BDE-47-pre-treated mononuclear cells of autistic children as compared to control children [91], suggesting that the former may respond differently to PBDEs.
Heavy Metals
Heavy metals, particularly lead (Pb) and mercury (Hg), are widespread environmental toxins that can produce multisystem damage. Fetal exposure through the placenta can result in impaired cognitive development and behavioral disturbances, such as low IQ, ADHD and impaired motor skills [92–94]. The relationship between heavy metal exposure and ASD is highly controversial. Contrasting results have been found with regard to serum levels of Pb and Hg in ASD children [95–98]. The debate on the possible role of the vaccine preservative thimerosal (composed by ethyl-mercury) is outlined in the next section. However, sources other than thimerosal could potentially determine chronic Hg exposure resulting in increased ASD risk, as suggested by ecological studies in areas with higher levels of Hg in ambient air [99, 100]. Furthermore, the descendants of a cohort of individuals with Hg hypersensitivity showed ASD prevalence above general population rates [101], again supporting the role of Gene x Environment interactions. Moreover, differences in gene transcription profiles correlated with Hg and Pb plasma levels were detected between blood samples of ASD and TD children, suggesting that some ASD children may display a unique susceptibility to heavy metals, presumably due to their peculiar genetic profile [102, 103]. Finally, the concomitant administration of Pb and proinflammatory cytokines altered the expression of metalloproteinase genes in glial cells and acted on neuronal tissue remodelling, while the administration of only one of the two agents did not, unveiling possible synergistic or permissive interactions between heavy metals and proinflammatory factors [104].
In summary, exposure to heavy metals from sources other than vaccines, combined with genetic variants promoting immune dysfunction or impairing neurodevelopmental processes, could potentially exert small additive effects in a limited number of ASD children. However, it should be considered that the elevated blood levels of heavy metals found in many autoimmune disorders, such as rheumatoid arthritis [105], stem from active dysimmunity rather than from environmental contamination, and that similarly active dysimmunity may well be present in a consistent subset of ASD children, for which metal chelation therapy appears absolutely unjustified.
Vaccines
In the late 1990s, the MMR (measles mumps and rubella) vaccine and thimerosal, an ethyl mercury preservative, began to be regarded as potential causal factors for ASD. Public concern rose following Andrew Wakefield’s paper, which connected MMR and autism [106], and the almost coincidental recommendation to remove thimerosal-containing vaccines from the market, made in 1999 by the American Academy of Pediatrics (AAP), jointly with Public Health Services. Huge research efforts have been made to clarify whether the suggested causal link is scientifically valid. Large national databases have been analyzed to take a deeper look at the effects of changes in vaccine policies on autism prevalence rates. Collectively, the majority of epidemiological studies have not found a relationship between autism prevalence and MMR or thimerosal exposure. For example, in the UK, autism prevalence increased between 1979 and 1992, but no "step-up" effect, or change in trend, was seen after the introduction of MMR in 1988 [107]. Moreover, autism incidence continued to rise after steady MMR coverage was obtained, indicating no correlation between the two [108]. A large retrospective cohort study of all Danish children born from January 1991 through December 1998 showed no increase in the risk of ASD for MMR vaccinated children [109]. No differences were found in rates of developmental regression and gastrointestinal symptoms before and after the introduction of the MMR vaccine, and between MMR-exposed and -unexposed children with autism [110]. Similar results were obtained in epidemiological studies addressing the role of thimerosal-containing vaccines (TCVs). A large Danish study investigated ASD incidence between 1971 and 2000: no increase was seen until 1990, although thimerosal had been in use for several years. Conversely, the rising trend of autism incidence, recorded starting in 1991, continued unchanged in spite of the removal of TCVs in 1992 [111]. Another population-based cohort study of all children born in Denmark between 1990 and 1996 also provided negative findings, showing a lack of dose–response association between thimerosal and autism severity [112]. Similarly, results from studies conducted in the UK and USA did not show increased autism risk in children exposed to TCVs, nor significant associations between TCVs and neurodevelopmental adverse outcomes [113–115]. Consequently, in 2002 the AAP withdrew its 1999 recommendation, and in 2004, the Institute of Medicine concluded that evidence favored the rejection of a link between the MMR vaccine or thimerosal and autism [116].
Despite the epidemiological evidence outlined above, the controversy on vaccine safety continues. Some have argued that thimerosal exposure has continued through influenza vaccines, recommended to pregnant women and infants, thus undermining epidemiological studies attempting to monitor the consequences of its removal after 1999. Animal models have shown that thimerosal may affect the CNS, especially during development: persistent abnormalities in brain monoaminergic system after prenatal exposure to thimerosal at E9 have been reported in mice [117, 118]; increased oxidative stress in cerebellar cells, as well as impaired motor learning and delayed startle response have been observed in rats prenatally and postnatally exposed to Et-Hg [119]. In vitro studies have found that Et-Hg increases oxidative stress, reduces GSH availability, and alters Ca2+ signaling [120, 121]. Even more interestingly, both in mice and rats, males and strains prone to develop autoimmunity appear more sensitive to thimerosal than females and strains resistant to autoimmunity [122–125]. A recent study on 11 families with an autistic child showed unaffected twins and non-twin siblings having thimerosal hypersensitivity in B lymphocytes in 4 of the 11 families, as indicated by elevated oxidative stress targeting mitochondria [126].
Collectively, these results indicate that vaccines and thimerosal per se do not cause autism in the vast majority of patients. Changes in ASD incidence trends should have reflected changes in vaccination policies, as promptly and faithfully as lung cancer incidence has followed trends in smoking habits. Ironically, by preventing substantial numbers of infectious diseases like rubella, vaccinations actually prevent many cases of autism [127]. Nonetheless, some genetically vulnerable individuals could conceivably suffer from the consequences of vaccination and thimerosal, as well as of several other incidental conditions activating the immune system and enhancing oxidative stress during early infancy (a common example being the recurrent ear infections so frequently affecting children later diagnosed with autism during the first 12–18 months of life). By hindering energy metabolism, vaccines, thimerosal, and infections could each provide small, additive contributions toward the precipitation of a “regressive” form of autism, which would have otherwise evolved spontaneously at a slower and more progressive pace. However, many purely genetic forms of autism, such as those due to NLGN3 mutations, also typically display a regressive onset. This clearly speaks against regression as necessarily stemming from an environmental factor acting postnatally at the time when behavioral alterations are first noticed. Rather, this could be the time when defective neural networks fail to come “on-line” and to support the acquisition of novel cognitive functions.
Conclusions
An increasing body of evidence is showing that environmental factors can variably increase autism risk through multiple mechanisms, namely deregulating gene expression, altering signal transduction or cytosolic calcium homeostasis, and inhibiting enzymatic activities critical to brain development and neural function, causing oxidative stress or neuroinflammation. The array of environmental agents potentially involved in ASD is broad, including several not discussed here such as prenatal and perinatal stressors [128, 129], prenatal infections boosting IL6 production [130••], and neuroactive compounds produced by gut bacteria [131–133]. Meanwhile, the well-established causal role demonstrated for pathogenic mutations and CNVs in many cases of autism imposes a balanced view. This multiplicity of environmental agents and pathways, paired with the multiplicity of common and rare genetic variants causing autism in some and merely conferring susceptibility in others, should help us abandon the unrealistic hope of finding the “one cause” of autism in every patient and to tolerate complexity, as it occurs in nature and in human disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Arlington: American Psychiatric Publishing; 2013.
Robinson EB, Koenen KC, McCormick MC, Munir K, Hallett V, Happé F, et al. Evidence that autistic traits show the same etiology in the general population and at the quantitative extremes (5 %, 2.5 %, and 1 %). Arch Gen Psychiatry. 2011;68(11):1113–21.
Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of Autism Spectrum Disorders - Autism and Developmental Disabilities Monitoring Network, 14 Sites, United States. MMWR Surveill Summ 2012; 61:1–19.
Elsabbagh M, Divan G, Koh YJ, Kim YS, Kauchali S, Marcín C, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012;5(3):160–79. doi:10.1002/aur.239. Review.
Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–55.
Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism. 2012;3:9. By applying quantitative genetics techniques to simplex and multiplex families, the authors are able for the first time to estimate the relative weight of common genetic variants, each conferring a small, additive effect. This contribution is larger in multiplex families, where there is evidence of assortative mating in parents. The complex interplay of common and rare genetic variants in association with the environment is critical to the expression of ASD.
Landrigan PJ. What causes autism? Exploring the environmental contribution. Curr Opin Pediatr. 2010;22:219–25.
Persico AM, Napolioni V. Autism genetics. Behav Brain Res. 2013;251:95–112. doi:10.1016/j.bbr.2013.06.012.
Bromley RL, Mawer GE, Briggs M, Cheyne C, Clayton-Smith J, García-Fiñana M, et al. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J Neurol Neurosurg Psychiatry. 2013;84:637–43.
Kjaer D, Christensen J, Bech BH, Pedersen LH, Vestergaard M, Olsen J. Preschool behavioral problems in children prenatally exposed to antiepileptic drugs - A follow-up study. Epilepsy Behav. 2013 Sep 30. doi:pii: S1525-5050(13)00456-3. 10.1016/j.yebeh.2013.08.033
Cohen MJ, Meador KJ, Browning N, May R, Baker GA, Clayton-Smith J, Kalayjian LA, Kanner A, Liporace JD, Pennell PB, Privitera M, Loring DW; for the NEAD study group Fetal antiepileptic drug exposure: Adaptive and emotional/behavioral functioning at age 6 years.Epilepsy Behav. 2013 Sep 5. doi:pii: S1525-5050(13)00406-X. 10.1016/j.yebeh.20
Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA. 2013;309:1696–703.
Rasalam AD, Hailey H, Williams JH, Moore SJ, Turnpenny PD, Lloyd DJ, et al. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev Med Child Neurol. 2005;47:551–5.
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–41.
Hill EJ, Nagel DA, O'Neil JD, Torr E, Woehrling EK, Devitt A, et al. Effects of lithium and valproic acid on gene expression and phenotypic markers in an NT2 neurosphere model of neural development. PLoS One. 2013;8:e58822.
Rout UK, Clausen P. Common increase of GATA-3 level in PC-12 cells by three teratogens causing autism spectrum disorders. Neurosci Res. 2009;64:162–9.
Miyazaki K, Narita N, Narita M. Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int J Dev Neurosci. 2005;23:287–97.
Ingram JL, Peckhan SM, Tisdale B, Rodier PM. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol Teratol. 2000;22:319–24.
Rinaldi T, Silberberg G, Markram H. Hyperconnectivity of local neocortical microcircuitry induced by prenatal exposure to valproic acid. Cereb Cortex. 2008;18:763–70.
Markram K, Rinaldi T, La Mendola D, Sandi C, Markram H. Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmacol. 2008;33:901–12.
Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci U S A. 2007;104:13501–6.
Schanen NC. Epigenetics of autism spectrum disorders. Hum Mol Genet. 2006;15(Spec No 2):R138–150.
Ben-David E, Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry 2012 Nov 13. doi: 10.1038/mp.2012.148. [Epub ahead of print]
Hallene KL, Oby E, Lee BJ, Santaguida S, Bassanini S, Cipolla M, et al. Prenatal exposure to thalidomide, altered vasculogenesis, and CNS malformations. Neuroscience. 2006;142:267–83.
Ema M, Ise R, Kato H, ONeda S, Hirata-Koizumi M, Singh AV, et al. Fetal malformations and early embryonic gene expression in cynomologus monkeys maternally exposed to thalidomide. Reprod Toxicol. 2010;29:49–56.
Bandim JM, Ventura LO, Miller MT, Almeida HC, Costa AE. Autism and Möbius sequence: an exploratory study of children in northeastern Brazil. Arq Neuropsiquiatr. 2003;61:181–5.
Miller MT, Ventura L, Strömland K. Thalidomide and misoprostol: Ophthalmologic manifestations and associations both expected and unexpected. Birth Defects Res A Clin Mol Teratol. 2009;85:667–76.
Dufour-Rainfray D, Vourc'h P, Tourlet S, Guilloteau D, Chalon S, Andres CR. Fetal exposure to teratogens: evidence of genes involved in autism. Neurosci Biobehav Rev. 2011;35:1254–65.
Ploeger A, Raijmakers ME, van der Maas HL, Galis F. The association between autism and errors in early embryogenesis: what is the causal mechanism? Biol Psychiatry. 2010;67:602–7.
Libbey JE, Sweeten TL, McMahon WM, Fujinami RS. Autistic disorder and viral infections. J Neurovirol. 2005;11:1–10.
Chess S, Fernandez P, Korn S. Behavioral consequences of congenital rubella. J Pediatr. 1978;93:699–703.
Ivarsson SA, Bjerre I, Vegfors P, Ahlfors K. Autism as one of several disabilities in two children with congenital cytomegalovirus infection. Neuropediatrics. 1990;21:102–3.
Sweeten TL, Posey DJ, McDougle CJ. Brief report: autistic disorder in three children with cytomegalovirus infection. J Autism Dev Disord. 2004;34:583–6.
Yamashita Y, Fujimoto C, Nakajima E, Isagai T, Matsuishi T. Possible association between congenital cytomegalovirus infection and autistic disorder. J Autism Dev Disord. 2003;33:455–9.
Engman ML, Lewensohn-Fuchs I, Mosskin M, Malm G. Congenital cytomegalovirus infection; the impact of cerebral cortical malformations. Acta Paediatr. 2010;99:1344–9.
Grandjean P, Landrigan PJ. Developmental neurotoxicity of industrial chemicals: a silent pandemic. Lancet. 2006;368:2167–78.
Rice D, Barone Jr S. Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108:511–33.
Akimoto H. Global air quality and pollution. Science. 2003;302:1716–9.
Sram RJ, Binkova B, Dejmek, Bobak M. Ambient air pollution and pregnancy outcomes: a review of the literature. Environ Health Perspect. 2005;113:375–82.
Hansen CA, Barnett AG, Pritchard. The Effect of Ambient Air Pollution during Early Pregnancy on Fetal Ultrasonic Measurements during Mid-Pregnancy. Environ Health Perspect. 2008;116:362–9.
Suglia SF, Gryparis A, Wright RO, Schwartz J, Wright RJ. Association of black carbon with cognition among children in a prospective birth cohort study. Am J Epidemiol. 2008;167:280–6.
Morales E, Julvez J, Torrent M, de Cid R, Guxens M, Bustamante M, et al. Association of early-life exposure to household gas appliances and indoor nitrogen dioxide with cognition and attention behavior in preschoolers. Am J Epidemiol. 2009;169:1327–36.
Kalkbrenner AE, Daniels JL, Chen JC, Poole C, Emch M, Morrissey J. Perinatal exposure to hazardous air pollutants and autism spectrum disorders at age 8. Epidemiology. 2010;21:631–41.
Volk HE, Hertz-Picciotto I, Delwiche L, Lurmann F, McConnell R. Residential proximity to freeways and autism in the CHARGE study. Environ Health Perspect. 2011;119:873–7.
Volk HE, Lurmann F, Penfold B, Hertz-Picciotto I, McConnell R. Traffic-related air pollution, particulate matter, and autism. JAMA Psychiatry. 2013;70:71–7.
Becerra TA, Wilhelm M, Olsen J, Cockburn M, Ritz B. Ambient air pollution and autism in Los Angeles County, California. Environ Health Perspect. 2013;121:380–6.
Block ML, Calderón-Garcidueñas L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009;32:506–16.
Genc S, Zadeoglulari Z, Fuss SH, Genc K. The adverse effects of air pollution on the nervous system. J Toxicol. 2012;2012:782462.
Block ML, Wu X, Pei Z, Li G, Wang T, Veronesi B, et al. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004;18:1618–20.
Pereyra-Muñoz N, Rugerio-Vargas C, Angoa-Pérez M, Borgonio-Pérez G, Rivas-Arancibia S. Oxidative damage in substantia nigra and striatum of rats chronically exposed to ozone. J Chem Neuroanat. 2006;31:114–23.
Thirtamara Rajamani K, Doherty-Lyons S, Bolden C, Willis D, Hoffman C, Gu H, et al. Prenatal and early-life exposure to high-level diesel exhaust particles leads to increased locomotor activity and repetitive behaviors in mice. Autism Res. 2013;6(4):248–57. doi:10.1002/aur.1287.
Sheng L, Ding X, Ferguson M, McCallister M, Rhoades R, Hood DB, et al. Prenatal polycyclic aromatic hydrocarbon exposure leads to behavioral deficits and downregulation of receptor tyrosine kinase, MET. Toxicol Sci. 2010;118:625–34. Prenatal exposure to benzo(a)pyrene downregulates the postnatal expression of the autism gene MET during synaptogenesis, reduces MET promoter activity, and results in behavioral deficits in novel object discrimination. This study nicely demonstrates a specific gene x environment interaction involving a common airborne pollutant and an established autism gene using animal and cellular models.
Bouayed J, Desor F, Rammal H, Kiemer AK, Tybl E, Schroeder H, et al. Effects of lactational exposure to benzo[alpha]pyrene (B[alpha]P) on postnatal neurodevelopment, neuronal receptor gene expression and behaviour in mice. Toxicology. 2009;259:97–106.
Herbstman JB, Tang D, Zhu D, Qu L, Sjödin A, Li Z, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, benzo[a]pyrene-DNA adducts, and genomic DNA methylation in cord blood. Environ Health Perspect. 2012;120:733–8.
Furlong CE, Holland N, Richter RJ, Bradman A, Ho A, Eskenazi B. PON1 status of farmworker mothers and children as a predictor of organophosphate sensitivity. Pharmacogenet Genomics. 2006;16:183–90.
Bouchard M, Chevrier J, Harley KG, Kogut K, Vedar M, Eskenazi B, et al. Prenatal exposure to organophosphate pesticides and IQ in 7-year-old children. Environ Health Perspect. 2011;119:1189–95.
Engel S, Wetmur J, Chen J, Zhu C, Barr D, Canfield R, et al. Prenatal exposure to organophosphates, paraoxonase 1, and cognitive development in childhood. Environ Health Perspect. 2011;119:1182–8.
Rauh V, Arunajadai S, Horton M, Perera F, Hoepner L, Barr D, et al. Seven-year neurodevelopmental scores and prenatal exposure to chlorpyrifos, a common agricultural pesticide. Environ Health Perspect. 2011;119:1196–201.
Rauh V, Perera F, Horton M, Whyatt R, Bansal R, Peterson BS, et al. Brain anomalies in children exposed prenatally to a common organophosphate pesticide. Proc Natl Acad Sci U S A. 2012;109:7871–6. Structural MRI study describing differences between 20 children prenatally exposed to chlorpyrifos and 20 controls (age 5.9-11.2 yrs). Prenatal chlorpyrifos (CPF) exposure was estimated on the basis of CPF concentrations measured in umbilical cord blood at birth. Main CPF effects include enlargement of several temporal, frontal, and postcentral regions, cuneus, and precuneus, along with various sex- and IQ-specific interactions.
Roberts EM, English PB, Grether JK, Windham GC, Somberg L, Wolff C. Maternal residence near agricultural pesticide applications and autism spectrum disorders among children in the California Central Valley. Environ Health Perspect. 2007;115:1482–9.
Flaskos J. The developmental neurotoxicity of organophosphorus insecticides: a direct role for the oxon metabolites. Toxicol Lett. 2012;209:86–93.
Stamou M et al. Neuronal connectivity as a convergent target for gene x environment interactions that confer risk for Autism Spectrum Disorders. Neurotoxicol Teratol. 2013. doi:10.1016/j.ntt.2012.12.001.
Shelton F, Hertz-Picciotto I, Pessah IN. Tipping the balance of autism risk: potential mechanisms linking pesticides and autism. Environ Health Perspect. 2012;120:944–51.
Betancourt AM, Burgess SC, Carr RL. Effect of developmental exposure to chlorpyrifos on the expression of neurotrophin growth factors and cell-specific markers in neonatal rat brain. Toxicol Sci. 2006;92:500–6.
Quattrocchi CC, Wannenes F, Persico AM, Ciafrè SA, D’Arcangelo G, Farace MG, et al. Reelin is a serine protease of the extracellular matrix. J Biol Chem. 2002;277:303–9.
D'Amelio M, Ricci I, Sacco R, Liu X, D’Agruma L, Persico AM, et al. Paraoxonase gene variants are associated with autism in North America, but not in Italy: possible regional specificity in gene-environment interactions. Mol Psychiatry. 2005;10:1006–16.
Gaita L, Manzi B, Sacco R, Lintas C, Altieri L, Persico AM, et al. Decreased serum arylesterase activity in Autism Spectrum Disorders. Psychiatry Res. 2010;180:105–13.
Mullen BR, Khialeeva E, Hoffman DB, Ghiani CA, Carpenter EM. Decreased reelin expression and organophosphate pesticide exposure alters mouse behaviour and brain morphology. ASN Neuro. 2012;5(1):e00106. doi:10.1042/AN20120060.
Goines PE, Ashwood P. Cytokine dysregulation in Autism Spectrum Disorders (ASD): Possible role of the environment. Neurotoxicol Teratol. 2012. doi:10.1016/j.ntt.2012.07.006.
Costa LG, Giordano G. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology. 2007;28(6):1047–67.
Tsukimori K, Morokuma S, Hori T, Takahashi K, Hirata T, Otera Y, et al. Characterization of placental transfer of polychlorinated dibenzo-p-dioxins, dibenzofurans and polychlorinated biphenyls in normal pregnancy. J Obstet Gynaecol Res. 2013;39:83–90.
Roze E, Meijer L, Bakker A, Van Braeckel KN, Sauer PJ, Bos AF. Prenatal exposure to organohalogens, including brominated flame retardants, influences motor, cognitive, and behavioral performance at school age. Environ Health Perspect. 2009;117:1953–8.
Gascon M, Fort M, Martínez D, Carsin AE, Forns J, Vrijheid M, et al. Polybrominated diphenyl ethers (PBDEs) in breast milk and neuropsychological development in infants. Environ Health Perspect. 2012;120:1760–5.
Eskenazi B, Chevrier J, Rauch SA, Kogut K, Harley KG, Johnson C, et al. In utero and childhood polybrominated diphenyl ether (PBDE) exposures and neurodevelopment in the CHAMACOS study. Environ Health Perspect. 2013;121:257–62.
Boas M, Feldt-Rasmussen U, Main KM. Thyroid effects of endocrine disrupting chemicals. Mol Cell Endocrinol. 2012;355:240–8.
Craig ZR, Wang W, Flaws JA. Endocrine-disrupting chemicals in ovarian function: effects on steroidogenesis, metabolism and nuclear receptor signaling. Reproduction. 2011;142:633–46.
Fair PA, Stavros HC, Mollenhauer MA, DeWitt JC, Henry N, Kannan K, et al. Immune function in female B(6)C(3)F(1) mice is modulated by DE-71, a commercial polybrominated diphenyl ether mixture. J Immunotoxicol. 2012;9:96–107.
Watanabe W, Shimizu T, Sawamura R, Hino A, Konno K, Kurokawa M. Functional disorder of primary immunity responding to respiratory syncytial virus infection in offspring mice exposed to a flame retardant, decabrominated diphenyl ether, perinatally. J Med Virol. 2010;82:1075–82.
Bradner JM, Suragh TA, Wilson WW, Lazo CR, Stout KA, Kim HM, et al. Exposure to the polybrominated diphenyl ether mixture DE-71 damages the nigrostriatal dopamine system: Role of dopamine handling in neurotoxicity. Exp Neurol. 2013;241:138–47.
Giordano G, Kavanagh TJ, Costa LG. Neurotoxicity of a polybrominated diphenyl ether mixture (DE-71) in mouse neurons and astrocytes is modulated by intracellular glutathione levels. Toxicol Appl Pharmacol. 2008;232:161–8.
Pessah IN, Cherednichenko G, Lein PJ. Minding the calcium store: Ryanodine receptor activation as a convergent mechanism of PCB toxicity. Pharmacol Ther. 2010;125:260–85.
Yang D, Kim KH, Phimister A, Bachstetter AD, Ward TR, Lein PJ, et al. Developmental exposure to polychlorinated biphenyls interferes with experience-dependent dendritic plasticity and ryanodine receptor expression in weanling rats. Environ Health Perspect. 2009;117:426–35.
Wayman GA, Yang D, Bose DD, Lesiak A, Ledoux V, Lein PJ, et al. PCB-95 promotes dendritic growth via ryanodine receptor-dependent mechanisms. Environ Health Perspect. 2012;120:997–1002. Golgi analysis of hippocampi from weanling rats prenatally exposed to PCB-95 via the maternal diet demonstrates that PCB-95 promotes dendritic overgrowth at concentrations as low as 2 pM. In culture, this effect is prevented by blocking ryanodine receptors (RyRs) either pharmacologically through FLA365 or using siRNA knockdown of RyRs.
Carayol J, Sacco R, Tores F, Rousseau F, Lewin P, Hager J, et al. Converging evidence for an association of ATP2B2 allelic variants with autism in males. Biol Psychiatry. 2011;70:880–7.
Palmieri L, Papaleo V, Porcelli V, Scarcia P, Gaita L, Sacco R, et al. Altered calcium homeostasis in autism-spectrum disorders: evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier AGC1. Mol Psychiatry. 2010;15:38–52. This study describes excessive aspartate/glutamate transport in postmortem autistic brains. This abnormality is not due to genetic mutations, but to excess in calcium levels, which overstimulates transporter activity. Oxidative stress is one of the ultimate consequences of this process.
Bal-Price AK, Coecke S, Costa L, Crofton KM, Fritsche E, Goldberg A, et al. Conference report: advancing the science of developmental neurotoxicity (DNT) testing for better safety evaluation. Altex-Altern Anim Ex. 2012;29:202–15.
Woods R, Vallero RO, Golub MS, Suarez JK, Ta TA, Yasui DH, et al. Long-lived epigenetic interactions between perinatal PBDE exposure and Mecp2308 mutation. Hum Mol Genet. 2012;21:2399–411.
Hertz-Picciotto I, Bergman A, Fängström B, Rose M, Krakowiak P, Pessah I, et al. Polybrominated diphenyl ethers in relation to autism and developmental delay: a case–control study. Environ Health. 2011;10:1.
Cheslack-Postava K, Rantakokko PV, Hinkka-Yli-Salomäki S, Surcel HM, McKeague IW, Kiviranta HA, et al. Maternal serum persistent organic pollutants in the Finnish Prenatal Study of Autism: A pilot study. Neurotoxicol Teratol. 2013;38:1–5. doi:10.1016/j.ntt.2013.04.001.
Mitchell MM, Woods R, Chi LH, Schmidt RJ, Pessah IN, Kostyniak PJ, et al. Levels of select PCB and PBDE congeners in human postmortem brain reveal possible environmental involvement in 15q11-q13 duplication autism spectrum disorder. Environ Mol Mutagen. 2012;53:589–98.
Ashwood P, Schauer J, Pessah IN, Van de Water J. Preliminary evidence of the in vitro effects of BDE-47 on innate immune responses in children with autism spectrum disorders. J Neuroimmunol. 2009;208:130–5.
Roegge CS, Schantz SL. Motor function following developmental exposure to PCBS and/or MEHG. Neurotoxicol Teratol. 2006;28:260–77.
Boucher O, Jacobson SW, Plusquellec P, Dewailly E, Ayotte P, Forget-Dubois N, et al. Prenatal methylmercury, postnatal lead exposure, and evidence of attention deficit/hyperactivity disorder among Inuit children in Arctic Québec. Environ Health Perspect. 2012;120(10):1456–61. doi:10.1289/ehp.1204976. Epub 2012 Aug 16.
Goodlad JK, Marcus DK, Fulton JJ. Lead and Attention-Deficit/Hyperactivity Disorder (ADHD) symptoms: A meta-analysis. Clin Psychol Rev. 2013;33:417–25.
Desoto MC, Hitlan RT. Blood levels of mercury are related to diagnosis of autism: a reanalysis of an important data set. J Child Neurol. 2007;22:1308–11.
Hertz-Picciotto I, Green PG, Delwiche L, Hansen R, Walker C, Pessah IN. Blood mercury concentrations in CHARGE Study children with and without autism. Environ Health Perspect. 2010;118:161–6.
Wright B, Pearce H, Allgar V, Miles J, Whitton C, Alderson-Day B, et al. A comparison of urinary mercury between children with autism spectrum disorders and control children. PLoS One. 2012;7(2):e29547.
Albizzati A, Morè L, Di Candia D, Saccani M, Lenti C. Normal concentrations of heavy metals in autistic spectrum disorders. Minerva Pediatr. 2012;64:27–31.
Blanchard KS, Palmer RF, Stein Z. The value of ecologic studies: mercury concentration in ambient air and the risk of autism. Rev Environ Health. 2011;26:111–8.
Palmer RF, Blanchard S, Wood R. Proximity to point sources of environmental mercury release as a predictor of autism prevalence. Health Place. 2009;15:18–24.
Shandley K, Austin DW. Ancestry of pink disease (infantile acrodynia) identified as a risk factor for autism spectrum disorders. J Toxicol Environ Health A. 2011;74:1185–94.
Stamova B, Green PG, Tian Y, Hertz-Picciotto I, Pessah IN, Sharp FR, et al. Correlations between gene expression and mercury levels in blood of boys with and without autism. Neurotox Res. 2011;19:31–48.
Tian Y, Green PG, Stamova B, Hertz-Picciotto I, Pessah IN, Sharp FR, et al. Correlations of gene expression with blood lead levels in children with autism compared to typically developing controls. Neurotox Res. 2011;19:1–13.
Lahat N, Shapiro S, Froom P, Kristal-Boneh E, Inspector M, Miller A. Inorganic lead enhances cytokine-induced elevation of matrix metalloproteinase MMP-9 expression in glial cells. J Neuroimmunol. 2002;132:123–8.
Wanchu A, Sud A, Bambery P, Prasad R, Kumar V. Plasma and peripheral blood mononuclear cells levels of Zn and Cu among Indian patients with RA. Ann Rheum Dis. 2002;61:88.
Wakefield AJ, Murch SH, Anthony A, Linnell J, Casson DM, Malik M, et al. Ileal-lymphoid-nodular hyperplasia, non-specific colitis, and pervasive developmental disorder in children. Lancet. 1998;351(9103):637–41.
Taylor B, Miller E, Farrington CP, Petropoulos MC, Favot-Mayaud I, Li J, et al. Autism and measles, mumps, and rubella vaccine: no epidemiological evidence for a causal association. Lancet. 1999;353(9169):2026–9.
Kaye JA, del Mar M-MM, Jick H. Mumps, measles, and rubella vaccine and the incidence of autism recorded by general practitioners: a time trend analysis. BMJ. 2001;322(7284):460–3.
Madsen KM, Hviid A, Vestergaard M, Schendel D, Wohlfahrt J, Thorsen P, et al. A population-based study of measles, mumps, and rubella vaccination and autism. N Engl J Med. 2002;347(19):1477–82.
Taylor B, Miller E, Lingam R, Andrews N, Simmons A, Stowe J. Measles, mumps, and rubella vaccination and bowel problems or developmental regression in children with autism: population study. BMJ. 2002;324(7334):393–6.
Madsen KM, Lauritsen MB, Pedersen CB, Thorsen P, Plesner AM, Andersen PH, et al. Thimerosal and the occurrence of autism: negative ecological evidence from Danish population-based data. Pediatrics. 2003;112(3 Pt 1):604–6.
Hviid A, Stellfeld M, Wohlfahrt J, Melbye M. Association between thimerosal-containing vaccine and autism. JAMA. 2003;290(13):1763–6.
Andrews N, Miller E, Grant A, Stowe J, Osborne V, Taylor B. Thimerosal exposure in infants and developmental disorders: a retrospective cohort study in the United Kingdom does not support a causal association. Pediatrics. 2004;114(3):584–91.
Verstraeten T, Davis RL, DeStefano F, Lieu TA, Rhodes PH, Black SB, et al. Safety of thimerosal-containing vaccines: a two-phased study of computerized health maintenance organization databases. Pediatrics. 2003;112(5):1039–48.
Heron J, Golding J. Thimerosal exposure in infants and developmental disorders: a prospective cohort study in the United kingdom does not support a causal association. Pediatrics. 2004;114(3):577–83.
Institute of Medicine (US) Immunization Safety Review Committee. Immunization Safety Review: Vaccines and Autism. Washington (DC): National Academies Press (US); 2004.
Ida-Eto M, Oyabu A, Ohkawara T, Tashiro Y, Narita N, Narita M. Embryonic exposure to thimerosal, an organomercury compound, causes abnormal early development of serotonergic neurons. Neurosci Lett. 2011;505(2):61–4. doi:10.1016/j.neulet.2011.05.053.
Ida-Eto M, Oyabu A, Ohkawara T, Tashiro Y, Narita N, Narita M. Prenatal exposure to organomercury, thimerosal, persistently impairs the serotonergic and dopaminergic systems in the rat brain: implications for association with developmental disorders. Brain Dev. 2013;35(3):261–4. doi:10.1016/j.braindev.2012.05.004.
Ueha-Ishibashi T, Oyama Y, Nakao H, Umebayashi C, Nishizaki Y, Tatsuishi T, et al. Effect of thimerosal, a preservative in vaccines, on intracellular Ca2+ concentration of rat cerebellar neurons. Toxicology. 2004;195(1):77–84.
Migdal C, Foggia L, Tailhardat M, Courtellemont P, Haftek M, Serres M. Sensitization effect of thimerosal is mediated in vitro via reactive oxygen species and calcium signaling. Toxicology. 2010;274(1–3):1–9. doi:10.1016/j.tox.2010.04.016.
Guzzi G, Pigatto PD, Spadari F, La Porta CA. Effect of thimerosal, methylmercury, and mercuric chloride in Jurkat T Cell Line. Interdiscip Toxicol. 2012;5(3):159–61. doi:10.2478/v10102-012-0026-1.
Branch DR. Gender-selective toxicity of thimerosal. Exp Toxicol Pathol. 2009;61(2):133–6. doi:10.1016/j.etp.2008.07.002.
Hornig M, Chian D, Lipkin WI. Neurotoxic effects of postnatal thimerosal are mouse strain dependent. Mol Psychiatry. 2004;9(9):833–45.
Sulkowski ZL, Chen T, Midha S, Zavacki AM, Sajdel-Sulkowska EM. Maternal thimerosal exposure results in aberrant cerebellar oxidative stress, thyroid hormone metabolism, and motor behavior in rat pups; sex- and strain-dependent effects. Cerebellum. 2012;11(2):575–86. doi:10.1007/s12311-011-0319-5.
Sharpe MA, Gist TL, Baskin DS. B-lymphocytes from a population of children with autism spectrum disorder and their unaffected siblings exhibit hypersensitivity to thimerosal. J Toxicol. 2013;2013:801517. doi:10.1155/2013/801517.
Sharpe MA, Livingston AD, Baskin DS. Thimerosal-Derived Ethylmercury Is a Mitochondrial Toxin in Human Astrocytes: Possible Role of Fenton Chemistry in the Oxidation and Breakage of mtDNA. J Toxicol. 2012;2012:373678. doi:10.1155/2012/373678. Epub 2012 Jun 28.
Berger BE, Navar-Boggan AM, Omer SB. Congenital rubella syndrome and autism spectrum disorder prevented by rubella vaccination–United States, 2001–2010. BMC Public Health. 2011;11:340. doi:10.1186/1471-2458-11-340.
Guinchat V, Thorsen P, Laurent C, Cans C, Bodeau N, Cohen D. Pre-, peri- and neonatal risk factors for autism. Acta Obstet Gynecol Scand. 2012;91:287–300.
Lyall K, Pauls DL, Spiegelman D, Ascherio A, Santangelo SL. Pregnancy complications and obstetric suboptimality in association with autism spectrum disorders in children of the Nurses' Health Study II. Autism Res. 2012;5:21–30.
Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26:607–16. Activation of the maternal immune system by injecting the viral mimic poly(I:C) in mice during pregnancy, produces behavioral abnormalities in the offspring pertaining to autism core symptoms in humans, namely abnormal social, communication and stereotypic behaviors. Previous work by the same group supports a central role for IL-6 in this effect.
Altieri L, Neri C, Sacco R, Curatolo P, Benvenuto A, Muratori F, et al. Urinary p-cresol is elevated in small children with Autism Spectrum Disorder. Biomarkers. 2011;16:252–60.
Persico AM, Napolioni V. Urinary p-cresol in autism spectrum disorder. Neurotoxicol Teratol. 2013;36:82–90.
MacFabe DF, Cain NE, Boon F, Ossenkopp KP, Cain DP. Effects of the entericbacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: Relevance to autism spectrum disorder. Behav Brain Res. 2011;217:47–54.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Antonio M. Persico has grants/grants pending with the Autism Research Institute.
Sara Merelli declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Persico, A.M., Merelli, S. Environmental Factors in the Onset of Autism Spectrum Disorder. Curr Dev Disord Rep 1, 8–19 (2014). https://doi.org/10.1007/s40474-013-0002-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40474-013-0002-2
Keywords
- Air pollution
- Autism
- Autism spectrum disorder
- Benzo(a)pyrene
- Environment
- Environmental factors
- Halogenated aromatic hydrocarbons
- Heavy metals
- Mercury
- Misoprostol
- MMR
- Organophosphates
- Pervasive Developmental Disorders
- PBDEs
- PCBs
- p-cresol
- Pesticides
- Polybrominated diphenyl ethers
- Polychlorinated biphenyls
- Thalidomide
- Thimerosal
- Vaccines
- Valproate