
Abstract
Background
Ciclosporin is used as an immunosuppressant in current clinical practice but recent research implies novel indications for the drug, such as neuro- and cardioprotection. The intravenous formulation currently on the market, Sandimmune® Injection (Sandimmune®), uses Cremophor® EL as emulsifying excipient. Cremophor® EL is known to cause hypersensitivity reactions in some patients, ranging from skin reactions to potentially fatal anaphylactic shock.
Objectives
The primary objective was to assess if CicloMulsion®, a Cremophor® EL-free lipid emulsion of ciclosporin for intravenous administration, is bioequivalent to Sandimmune®, and the secondary objective was to compare the tolerability profiles of the two preparations.
Methods
This was a single-centre, open-label, subject-blind, laboratory-blind, single-dose, randomized, two-treatment, two-period, two-sequence crossover study of the pharmacokinetics of two formulations of intravenous ciclosporin. Fifty-two healthy volunteer subjects were administered 5 mg/kg of each of the two formulations of ciclosporin as a 4-h intravenous infusion. The last blood sample was acquired 48 h after the end of the infusion. Bioequivalence assessments according to current guidelines were performed.
Results
The geometric mean ratios for CicloMulsion®/Sandimmune® (90 % confidence interval [CI]) were 0.90 (0.88, 0.92) for AUC0–last (area under the blood concentration–time curve from time zero to time of last measurable concentration) and 0.95 (0.92, 0.97) for Cmax (maximum blood concentration). For all additional variables analysed, the 90 % CIs were also within the accepted bioequivalence range of 0.80–1.25. One anaphylactoid and one anaphylactic reaction, both classified as serious adverse events, were reported after treatment with Sandimmune®. No serious adverse events were recorded after treatment with CicloMulsion®.
Conclusion
We have assessed the pharmacokinetics and tolerability of a new Cremophor® EL-free lipid emulsion of ciclosporin, CicloMulsion®, compared to Sandimmune®. The proportion of adverse events was significantly higher for the Cremophor® EL-based product Sandimmune®. We conclude that CicloMulsion® is bioequivalent to Sandimmune® and exhibits fewer adverse reactions.
Similar content being viewed by others


1 Introduction
Ciclosporin, discovered in 1969 and first registered for pharmacological use in the early 1980s, is a polypeptide derived from the fungus species Tolypocladium inflatum. It binds to cyclophilins, a family of peptidylprolyl isomerases, that function as molecular chaperones. In current clinical practice ciclosporin is utilized as an immunosuppressant, reducing the T-lymphocyte function via inhibition of calcineurin by the ciclosporin–cyclophilin A complex [1]. Indications include solid organ or bone marrow transplantation and treatment of autoimmune conditions such as psoriasis, atopic dermatitis, pyoderma gangrenosum, ulcerative colitis, idiopathic nephrotic syndrome and inflammatory uveitis [2–8]. Further, ciclosporin binding to cyclophilin D in the mitochondrial matrix stabilizes membrane proteins participating in mitochondrial permeability transition, protecting the organelle from negative effects of oxidative stress and calcium overload. Ciclosporin is thus a promising therapeutic agent in conditions related to mitochondrial dysfunction such as traumatic brain injury, myocardial infarction and neurodegenerative disorders [9–13]. The molecule is highly hydrophobic and requires a lipophilic solvent for administration. The intravenous (IV) form of ciclosporin currently on the market, Sandimmune® Injection (Novartis Pharma Stein AG, Switzerland) [Sandimmune®], is a concentrate for infusion containing ethanol and polyoxyethylated castor oil, Cremophor® EL (CrEL).
CrEL is not inert [14] and there have been a number of reports of serious adverse effects after administration of IV ciclosporin due to reactions to this carrier medium [15–22]. Hypersensitivity reactions to CrEL have also been reported when used for other IV preparations of drugs such as the anxiolytic agent diazepam and the anaesthetic drug Althesin®, a combination of alphaxolone and alphadolone [23–28]. In rodents, neurotoxicity, cardiotoxicity and nephrotoxicity due to IV ciclosporin dissolved in CrEL have been demonstrated [29–32] and, in a canine model, CrEL has been shown to reduce cardiac output and hepatic blood flow in a non-dose-dependent fashion [33]. The effects of CrEL include complement activation, histamine release and severe hypersensitivity reactions [14, 23, 25, 27].
Ciclosporin in CrEL requires a dilution step prior to administration. Improper preparation of CrEL-containing formulations has been reported to cause anaphylactoid reactions [34, 35]. An additional concern with the use of ethanol and CrEL is the leaching of plasticizers from polyvinyl chloride (PVC) bags and infusion sets used in routine clinical practice. Consequently, preparation and administration should be done using glass or other non-PVC infusion sets [36, 37].
Concern about the safety of CrEL as a carrier medium for IV drugs has been raised on numerous occasions and several drugs that previously were produced in preparations with CrEL are now available only with other carrying media such as lipid emulsions. Known examples are propofol [23, 38] and diazepam [39]. Others, such as the chemotherapeutic agent paclitaxel, are available both with and without CrEL as solvent [40], and the anaesthetic drug Althesin® that contained CrEL is no longer marketed for use in humans [23, 27, 41].
In this study (ClinicalTrials.gov identifier NCT01692834), the objective was to assess the pharmacokinetics of a novel CrEL-and ethanol-free ready-to-use preparation of ciclosporin for IV administration, CicloMulsion® (NeuroVive Pharmaceutical AB, Lund, Sweden), also known as NeuroSTAT®, in relation to the CrEL-containing product currently on the market (Sandimmune® Injection) and to assess whether the two formulations are bioequivalent. A ready-to-use preparation without CrEL potentially offers increased patient safety with fewer adverse events due to improper handling, dilution or reactions to CrEL.
2 Methods
2.1 Study Design
This was a single-centre, open-label, subject-blind, laboratory-blind, single-dose, randomized, two-treatment, two-period, two-sequence crossover study of the pharmacokinetics of two formulations of IV ciclosporin. The primary objective was to assess the pharmacokinetics, and the secondary objective was to compare the tolerability profiles of the two preparations.
The study protocol, including amendments, subject information sheets and informed consent documents, were reviewed by the Ethics Committee of the Faculty of Health Sciences of the University of the Free State (Reference number ETOVS 65/09), and by the South African Medicines Control Council (Reference number BE 2009009), and written approval was acquired. The study was performed in accordance with the declaration of Helsinki and Guideline for Good Clinical Practice issued by the International Conference on Harmonization. It was designed to comply with the Guidance for Industry—Statistical Approaches to Establishing Bioequivalence issued by the United States Department of Health and Human Services, Food and Drug Administration (FDA) [42], and the Note for Guidance on the Investigation of Bioavailability and Bioequivalence by The European Medicines Agency (EMA) [43].
2.2 Ciclosporin Formulations
The reference formulation of ciclosporin used was Sandimmune® Injection (Novartis Pharma Stein AG, 50 mg/mL ciclosporin, United States Pharmacopeia, USP) containing Cremophor® EL (each mL of infusion concentrate was diluted in 20 mL 0.9 % saline solution prior to use). The test product used was CicloMulsion® (NeuroVive Pharmaceutical AB, Lund, Sweden) 5 mg/mL ready-to-use Cremophor- and ethanol-free ciclosporin Ph Eur/USP lipid emulsion. The lipid emulsion carrier is equivalent to the marketed product Lipovenoes® MCT 20 % (Fresenius Kabi AG, Graz, Austria) and each mL contains 100 mg of refined soya bean oil, 100 mg of medium-chain triglycerides, 12 mg of egg lecithin, 25 mg of glycerol, water, and sodium oleate and sodium hydroxide for pH adjustment.
2.3 Participants
Healthy male and healthy, non-pregnant, non-lactating female volunteers between 18 and 55 years of age with a body mass index within the range of 19–33 kg/m2 were eligible. Further inclusion criteria were body mass 60–100 kg, normal 12-lead electrocardiogram (ECG) and vital signs, clinically acceptable findings in medical history and physical examinations, laboratory results within the reference ranges (unless the deviation was considered irrelevant for the purpose of the study), willingness to undergo pre-, interim- and post-study physical examinations and laboratory investigations, ability to comprehend and willingness to sign statement of informed consent, and abstinence from tobacco during and 3 months prior to study. Female participants of childbearing age underwent a pregnancy test prior to each ciclosporin dosing and, if positive, were excluded from the study. During the study period, reliable, non-hormonal methods of contraception had to be used.
Exclusion criteria included evidence of psychiatric disorder, history of or current abuse of drugs (including alcohol), use of any medication within 2 weeks prior to first administration of study medication, participation in another study with an experimental drug with administration within 12 weeks prior to the current study, major illness during the last 3 months, donation or loss of blood exceeding 500 mL during the 8 weeks before the first administration of the study drug, positive test for hepatitis B or C or HIV, positive urine drug screen, vaccination of any kind within 4 weeks of first dose or planning vaccination within 3 months of last dose, close family member receiving live vaccine during study or within 3 months post-study, and hypotension or hypertension during screening period. History of any of the following diseases was also a criterion for exclusion from the study: any type of malignancy, immunodeficiency, tendency toward recurrent infections, known untreated parasitic infection, allergy to any compound in the reference and test product, or to egg or soya bean, any bronchospastic diseases, epilepsy, porphyria, psoriasis, atopic dermatitis, hypercholesterolaemia, gout, rheumatoid arthritis or kidney disease. Care was taken to include both female and male participants of both Caucasian and non-Caucasian race.
Written informed consent was obtained from all participants before study enrollment.
2.4 Sampling Period
Subjects were randomized into two treatment sequences: the test product followed by the reference product or vice versa. There was a washout period set to 14–21 days between the first and second treatment period. Participants reported to the clinic the night before treatment for laboratory testing, including blood samples, pregnancy test and urine drug screen. The subjects were instructed not to ingest any citrus fruits and/or apple or pineapple 72 h prior to start of infusion and, within 24 h, no alcohol or any caffeine-containing products were permitted. On the clinic day, the only food served before drug administration was a standardized breakfast. Through an indwelling IV cannula, the subjects received either 5 mg/kg CicloMulsion® (test) or 5 mg/kg Sandimmune® (reference), infused at a constant rate over 4 h with a syringe pump. The dose recommended for induction of immunosuppression with Sandimmune® in clinical practice is 3–6 mg/kg/day. The same arm was used for administration during both treatment periods. All infusion equipment was compatible with both the reference and the test product. Through an IV cannula in the contra-lateral arm, a total of 22 blood samples for ciclosporin analysis were obtained pre-dose and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 7, 8, 10, 14, 18, 24, 30, 36 and 48 h after start of infusion. All samples were collected in vials containing ethylenediaminetetraacetic acid (EDTA), labeled and stored at −70 °C until analysis. Whole blood ciclosporin-concentration was assayed with liquid chromatography-tandem mass spectrometry. The method was validated according to current FDA guidelines [44]. The lower limit of quantification (LLQ) was 39.39 ng/mL and the mean coefficient of variation (CV) was 3.5 % for ciclosporin. Complete sets of calibration standards and quality controls were included within each run.
Subjects were allowed to leave the clinic 24 h after start of administration of study medication, provided they returned for the subsequent collection of blood samples. Vital signs were monitored during the infusion and recorded 2 h after completion of infusion and after the last blood sample was drawn. Meals and drinks during clinic days were standardized. Water was allowed ad libitum before and after infusion of study medication. Alcohol, caffeine, citrus fruit, apple and pineapple were not allowed until the last blood sample was drawn in each treatment period.
2.5 Tolerability Assessment
Each subject was carefully monitored for adverse events (AEs) during infusion and was questioned on the study day for any symptoms of such events. AEs were graded as mild, moderate or severe according to the following definitions:
Mild: Causing no limitation of usual activities; the subject may experience slight discomfort.
Moderate: Causing some limitation of usual activities; the subject may experience annoying discomfort.
Severe: Causing inability to carry out usual activities; the subject may experience intolerable discomfort or pain.
The investigator judged each AE with regard to causality to the administered medical product as “certain,” “probable,” “possible,” “unlikely,” “not related” or “not assessable”. Every AE was coded with the Medical Dictionary for Regulatory Activities (MedDRA) and reported according to strict criteria. The proportions of overall AEs and AEs per organ class were compared between CicloMulsion® and Sandimmune® by means of 95 % confidence interval (CI) for the difference between paired proportions and p values from McNemar’s test.
2.6 Pharmacokinetics and Bioequivalence Assessment
Pharmacokinetic variables for ciclosporin were calculated by use of non-compartmental methods using WinNonlin® Professional version 5.2 (Pharsight Corporation, Mountain View, CA, USA). Statistical analysis was made using SAS® Software version 9.1 (SAS institute, Cary, NC, USA). All values below LLQ prior to the first positive sample were substituted with zero. For the pharmacokinetic assessment, the terminal values below the LLQ were ignored.
CicloMulsion® was compared to Sandimmune® with respect to a number of pharmacokinetic variables using analysis of variance (ANOVA) with sequence, subject within sequence, product and period effects on log-transformed data. The parameters compared were area under the blood concentration–time curves (AUCs) from time zero to time of last measurable concentration (AUC0–last), time zero to infinity (AUC0–∞), time 4 h to infinity (AUC4–∞), time zero to time 4 h (AUC0–4) and time 4 h to time of last measurable concentration (AUC4–last), maximum blood ciclosporin concentration (Cmax), apparent terminal half-life (t½β), clearance (CL) and mean residence time (MRT). Cmax, AUC0–∞, AUC0–last and AUC4–∞ were considered primary variables and the remainder secondary variables. Point estimates and 90 % CI for the CicloMulsion®/Sandimmune® geometric mean ratios of all variables were calculated. The two products were considered bioequivalent if the 90 % CI for the primary variables fell within the limits of 0.8 and 1.25.
Based on the FDA and EMA recommended bioequivalence range of 0.80–1.25 for Cmax, AUC0–last and AUC0–∞, an estimated within-subject CV of 35 %, and a test/reference mean ratio between 0.95 and 1.05, 52 subjects were needed to achieve a power of 80 % at an alpha level of 0.05 to show bioequivalence [45].
2.7 Modifications in Study Design
The initial study design did not include any premedication, but, due to an unexpectedly high incidence of serious adverse events (SAEs) to the reference product, the remainder of the study was performed with premedication. An amendment to the study protocol was written and approved by the ethics committees mentioned above. For the sake of consistency, premedication was used prior to infusion of both CicloMulsion® and Sandimmune® even though the AEs triggering the initiation of premedication were observed only following Sandimmune® administration. Thirteen subjects completed both treatment periods without premedication. Eighteen subjects received the test product in the first treatment period without premedication, and received the reference product with premedication in the second treatment period. The remainder (21 subjects) received premedication during both treatment periods, and thus with both the test and reference product.
The premedication consisted of one 50 mg capsule of diphenhydramine orally 1 h prior to commencement of infusion of test or reference drug, dexamethasone 10 mg by slow IV injection, and ranitidine 50 mg IV infusion over 5 min approximately 30 min prior to each dosing of study drug. The study was put on hold for the protocol amendments to be approved. This caused the washout period for the 18 subjects who received the test product in the first treatment period without premedication and the reference product with premedication in the second treatment period to be prolonged to more than 6 weeks. None of the premedications used are known to change the pharmacokinetic properties of ciclosporin or affect the bioanalytical assay.
3 Results
Sixty-five volunteers were enrolled in the study and randomized to a treatment sequence. Two subjects were withdrawn before first dosing due to illness, and 11 more withdrew before completion. Of these 11, three were due to consent withdrawal, two due to investigator/sponsor decision (uncertainty of dose received due to problems with infusion), and six due to AEs. None of these subjects were included in the bioequivalence analysis. Fifty-two participants completed the study and were included in the pharmacokinetic evaluation. The demographics of the subjects completing the study are presented in Table 1.
Sixty-three participants received at least one dose of study medication and were monitored for adverse reactions. Due to an unexpectedly high number of serious adverse reactions to Sandimmune®, the study protocol was changed and premedication as described above was introduced. The statistical analysis of incidence of AEs was performed solely including the 13 participants who received both the test and reference drug without premedication, making it less conclusive.
3.1 Pharmacokinetics and Bioequivalence Assessment
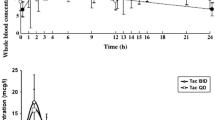
For each time point, arithmetic means with standard deviation were calculated for the whole blood ciclosporin-concentrations for the test drug CicloMulsion® and the reference drug Sandimmune®. A graphical presentation of the ciclosporin concentrations over time is provided in Fig. 1. In Fig. 2a, b, concentration–time curves are presented separately for subjects receiving and not receiving concomitant premedication. The pharmacokinetic parameters are presented in Table 2. The part of the AUC0–∞ that was extrapolated was approximately 5 % for both of the study medications, indicating that a reliable estimate of the AUC0–∞ was obtained. No values below LLQ were present between positive samples.

Arithmetic mean ± SD of blood concentration of ciclosporin in participants administered CicloMulsion® or Sandimmune® (n = 52)

Arithmetic mean ± SD of blood concentration of ciclosporin in participants administered a CicloMulsion® or b Sandimmune®, with or without premedication consisting of diphenhydramine 50 mg orally, dexamethasone 10 mg intravenously and ranitidine 50 mg intravenously
The statistical analysis of bioequivalence after dosing with CicloMulsion® and Sandimmune® is presented in Table 3. The point estimates of the CicloMulsion®/Sandimmune® geometric mean ratios (90 % CI) of the primary variables Cmax and AUC0–last for ciclosporin were 0.95 (0.92, 0.97) and 0.90 (0.88, 0.92), respectively, and AUC0–∞ and AUC4–∞ for ciclosporin were 0.90 (0.88, 0.92) and 0.87 (0.84, 0.90), respectively. Thus, the 90 % CIs of all primary variables were within the range acceptable for bioequivalence of 0.80–1.25. The 90 % CI for all secondary variables also met the criteria for bioequivalence (Table 3).
3.2 Tolerability
Out of 63 subjects, 55 reported one or more AEs. AEs graded as at least “possibly” related to the study medication were predominantly reported from the medDRA-coded organ classes of nervous system disorders and vascular disorders (Table 4). The nervous system disorders included headache, burning sensation, paraesthesia, dizziness and sensory loss. The vascular disorders included events of hot flushes and orthostatic hypotension. When subjects received Sandimmune® without premedication, 84 % (16/19) experienced AEs assessed as at least possibly related to the study medication, compared to 64 % (21/33) when they received CicloMulsion®. With premedication, the figures were 76 % (31/41) for Sandimmune® and 67 % (16/24) for CicloMulsion®. Proportional analysis of AEs was performed for the subjects treated with both CicloMulsion® and Sandimmune® without premedication. There was a significantly higher proportion of overall AEs (p = 0.003) and vascular disorders (p = 0.03) when subjects were treated with Sandimmune®. No other proportions were significantly different between the two formulations.
Two SAEs were recorded. The first affected a male, aged 21 years, who in the first treatment period was randomized to receive the reference medication, Sandimmune®. Directly after the start of infusion he experienced shortness of breath and a flushing sensation. Infusion was halted, treatment with 40 % oxygen via Hudson mask was initiated, and he received promethazine 25 mg and hydrocortisone 200 mg IV. Due to continuing dyspnoea, inhalation with nebulized salbutamol was commenced. The event was considered resolved 37 min after the start of infusion and oxygen therapy was discontinued. The next day, he left the clinic but reported feeling tired for another 3 days. The event was diagnosed as an anaphylactoid reaction.
The second of the two SAEs reported regarded a 42-year-old male who also received the reference drug Sandimmune® in the first treatment period. Eleven minutes after infusion of the medication commenced he presented with coughing, facial flushing and dyspnoea. Chest auscultation revealed wheezing sounds and the peripheral blood oxygen saturation (SpO2) measured via pulse oximeter was 95 %. His condition quickly deteriorated, he became pale and sweaty, and a systolic/diastolic blood pressure of 75/42 mmHg was recorded. The infusion was stopped and therapy with 40 % oxygen via Hudson mask and IV infusion with Ringer lactate commenced. Blood pressure was not rising and the SpO2 dropped to 91 % after a couple of minutes; he was treated with 0.5 mg adrenaline and 25 mg promethazine intramuscularly and 5 mg salbutamol as nebulized inhalation. An infusion of 200 mg hydrocortisone also commenced. His blood pressure was normalized 5 min after the first medical intervention; within an hour all drug treatment could be discontinued. After 24 h, treatment with IV fluids was halted and the subject could leave the clinic. He reported feeling fatigued for another 5 days but could carry out all normal activities. The event was diagnosed as an anaphylactic reaction. Both subjects were excluded from the remainder of the study.
Due to the events described above, as well as an allergic reaction considered moderate and possibly related to Sandimmune® that also caused withdrawal of the subject, the study was put on hold and an amendment to the study protocol made with the addition of premedication as described above.
Three other subjects did not complete the study due to AEs. These were all moderate and possibly or probably related to the study medication. Out of these, two participants received the test product with premedication and one the reference product with premedication. No subjects receiving the test drug CicloMulsion® without premedication experienced AEs causing withdrawal from the study.
A summary of all AEs at least possibly related to the study medication is presented in Table 4. There were no clinically significant or consistent changes in laboratory values or ECG-findings due to ciclosporin administration. Vital signs and clinical findings remained essentially unchanged during the study for all participants except for the two SAEs described above.
4 Discussion
After a single IV dose of 5 mg/kg, CicloMulsion® and Sandimmune® met the conventional criteria for bioequivalence. CicloMulsion® was the better tolerated of the two ciclosporin formulations.
Three patients had to be excluded from the study due to adverse reactions to Sandimmune®; hence the study design was changed after a decision from local authorities. Due to the high incidence of SAEs in subjects receiving the CrEL-based product, premedication was introduced. The calculated pharmacokinetic parameters between subjects who did and did not receive premedication were similar, and bioequivalence could be established with a low within-subject CV (for most variables under 10 %) including premedicated and non-premedicated subjects. Thus, the changes in protocol and the introduction of premedication did not affect the pharmacokinetic profile of ciclosporin. For the assessment of tolerability, the change of the study protocol was not unproblematic. The amendments were introduced due to an unexpected high incidence of SAEs attributed to the CrEL-containing reference product, but for consistency in the pharmacokinetic evaluation, premedication was also given to subjects receiving the test product. This made the tolerability assessment less stringent since only 13 subjects received both drugs without premedication and could be included in the statistic evaluation. To draw any definite conclusions about tolerability, randomized clinical trials are warranted.
In clinical practice, most CrEL-containing IV drugs (such as the anti-cancer agent paclitaxel) are administered with premedication due to the known risk of hypersensitivity reactions to CrEL or, in the case of Sandimmune®, are often given as a part of a combined regimen with corticosteroids. It is feasible to assume that the actual incidence of adverse reactions to Sandimmune® is obscured by the protective effect of the corticosteroids.
A number of the reports of CrEL-reactions have been explained by improper dilution of the Sandimmune® Injection concentrate. CrEL has a greater specific gravity than water and a high viscosity and, unless properly mixed, will not be equally partitioned in the infusion bottle. Concentrations of CrEL and ciclosporin up to nine times higher than the intended dose have been reported during the first 10 min of infusion when mixed improperly [21, 34, 35]. With a stable, ready-to-use preparation without the need for dilution, this would not be an issue.
When Althesin® was withdrawn in the late 1980 s because of serious side effects due to CrEL, some authors argued that CrEL should not be used as a solvent for future drugs [46]. Until now, ciclosporin for IV administration has not been available with any other emulsifying excipient; this is surprising considering the extensive literature reporting serious or even fatal CrEL-related reactions [34, 47]. Even though all pharmacokinetic parameters evaluated in this study were well within the range of conventional criteria for bioequivalence, the authors suggest that blood ciclosporin levels should be monitored in patients where a target blood concentration is required.
Presently, ciclosporin is used mainly after solid organ or bone marrow transplantations and in the treatment of autoimmune conditions, but there are other potential indications for the molecule. Ciclosporin® is currently being investigated as pharmacological therapy in patients undergoing intervention treatment after myocardial infarction (the CIRCUS Phase III study, “Cyclosporine and Prognosis in Acute Myocardial Infarction [MI] Patients”, clinicaltrials.gov identifier NCT01502774). Phase II trials in traumatic brain injury with ciclosporin have been conducted and further clinical trials are under way using CicloMulsion® / NeuroSTAT® [10–13, 48]. For both of these patient categories, corticosteroids or antihistamines are not given as a part of standard therapy, possibly increasing the risk of allergic reactions to CrEL, and hence, a better tolerated CrEL-free formulation of ciclosporin would be advantageous.
There are several previous studies comparing pharmacokinetics of orally administered ciclosporin-formulations, but few for IV administration. In its oral form, ciclosporin has frequently been found to display a large variation in bioavailability between individuals [49–53] due to a number of factors, such as rate of gastric emptying, the rate of biliary, pancreatic and intestinal secretion, polymorphism in cytochrome P4503A enzymes, and different haplotypes of p-glycoprotein expressed in the gut wall mucosa. The intraindividual differences are usually explained by dietary factors and clinical condition [50–52, 54, 55].
There are examples of IV drugs in which the pharmacokinetic profiles have been significantly altered when a lipid emulsion was introduced as an emulsifier; known examples are propofol and diazepam [23, 56–58]. Taking this into account, the estimated within-subject CV was estimated to be 35 % when designing the study. It turned out to be under 10 % for all primary variables, supporting the view that most of the within-subject variation in bioavailability is due to factors primarily relevant to the orally administered formulations of ciclosporin. The interindividual CV % in this study was between 10 and 20 % for Cmax, AUC0–last and AUC0–∞, consistent with previous reports for IV ciclosporin [49, 59, 60]. The ongoing debate about the switchability of brand and generic formulations of orally administered ciclosporin for reasons of variability should therefore not be extrapolated to IV formulations of the drug.
5 Conclusion
From the study, we conclude that CicloMulsion®, a novel ciclosporin lipid emulsion developed for IV use, is bioequivalent to Sandimmune® Injection, and that this ethanol- and Cremophor® EL-free, ready-to-use formulation appears to be better tolerated.
References
Giorgio V, Soriano ME, Basso E, et al. Cyclophilin D in mitochondrial pathophysiology. Biochim Biophys Acta. 2010;1797(6–7):1113–8.
Griveas I, Visvardis G, Papadopoulou D, et al. Effect of cyclosporine therapy with low doses of corticosteroids on idiopathic nephrotic syndrome. Artif Organs. 2010;34(3):234–7.
Warren RB, Griffiths CE. Systemic therapies for psoriasis: methotrexate, retinoids, and cyclosporine. Clin Dermatol. 2008;26(5):438–47.
Fritsche L, Dragun D, Neumayer HH, et al. Impact of cyclosporine on the development of immunosuppressive therapy. Transpl Proc. 2004;36(2 Suppl):130S–4S.
Naganuma M, Fujii T, Watanabe M. The use of traditional and newer calcineurin inhibitors in inflammatory bowel disease. J Gastroenterol. 2011;46(2):129–37.
Hijnen DJ, ten Berge O, Timmer-de Mik L, et al. Efficacy and safety of long-term treatment with cyclosporin A for atopic dermatitis. J Eur Acad Dermatol Venereol. 2007;21(1):85–9.
Kacmaz RO, Kempen JH, Newcomb C, et al. Cyclosporine for ocular inflammatory diseases. Ophthalmology. 2010;117(3):576–84.
Storb R, Antin JH, Cutler C. Should methotrexate plus calcineurin inhibitors be considered standard of care for prophylaxis of acute graft-versus-host disease? Biol Blood Marrow Transpl. 2010;16(1 Suppl):S18–27.
Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50(7–8):983–97.
Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359(5):473–81.
Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16(9):783–92.
Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19(4):443–51.
Veech RL, Valeri CR, VanItallie TB. The mitochondrial permeability transition pore provides a key to the diagnosis and treatment of traumatic brain injury. IUBMB Life. 2012;64(2):203–7.
Dorr RT. Pharmacology and toxicology of Cremophor EL diluent. Ann Pharmacother. 1994;28(5 Suppl):S11–4.
Chapuis B, Helg C, Jeannet M, et al. Anaphylactic reaction to intravenous cyclosporine. N Engl J Med. 1985;312(19):1259.
Kahan BD, Wideman CA, Flechner S, et al. Anaphylactic reaction to intravenous cyclosporin. Lancet. 1984;1(8367):52.
Leunissen KML, Waterval PWG, Vanhooff JP. Anaphylactic reaction to intravenous cyclosporin. Lancet. 1985;1(8429):636.
Friedman LS, Dienstag JL, Nelson PW, et al. Anaphylactic reaction and cardiopulmonary arrest following intravenous cyclosporine. Am J Med. 1985;78(2):343–5.
Volcheck GW, Van Dellen RG. Anaphylaxis to intravenous cyclosporine and tolerance to oral cyclosporine: case report and review. Ann Allergy Asthma Immunol. 1998;80(2):159–63.
van Hooff JP, Bessems P, Beuman GH, et al. Absence of allergic reaction to cyclosporin capsules in patient allergic to standard oral and intravenous solution of cyclosporin. Lancet. 1987;2(8573):1456.
Mackie FE, Umetsu D, Salvatierra O, et al. Pulmonary capillary leak syndrome with intravenous cyclosporin A in pediatric renal transplantation. Pediatr Transpl. 2000;4(1):35–8.
Howrie DL, Ptachcinski RJ, Griffith BP, et al. Anaphylactoid reactions associated with parenteral cyclosporine use: possible role of Cremophor EL. Drug Intell Clin Pharm. 1985;19(6):425–7.
Baker MT, Naguib M. Propofol: the challenges of formulation. Anesthesiology. 2005;103(4):860–76.
Dye D, Watkins J. Suspected anaphylactic reaction to Cremophor EL. Br Med J. 1980;280(6228):1353.
Huttel MS, Schou Olesen A, Stoffersen E. Complement-mediated reactions to diazepam with Cremophor as solvent (Stesolid MR). Br J Anaesth. 1980;52(1):77–9.
Moneret-Vautrin DA, Laxenaire MC, Viry-Babel F. Anaphylaxis caused by anti-Cremophor EL IgG STS antibodies in a case of reaction to althesin. Br J Anaesth. 1983;55(5):469–71.
Briggs LP, Clarke RS, Watkins J. An adverse reaction to the administration of disoprofol (Diprivan). Anaesthesia. 1982;37(11):1099–101.
Ramsay MA, Savege TM, Simpson BR, et al. Controlled sedation with alphaxalone–alphadolone. Br Med J. 1974;2(5920):656–9.
Thiel G, Hermle M, Brunner FP. Acutely impaired renal function during intravenous administration of cyclosporine A: a cremophore side-effect. Clin Nephrol. 1986;25(Suppl 1):S40–2.
Verani R. Cyclosporine nephrotoxicity in the Fischer rat. Clin Nephrol. 1986;25(Suppl 1):S9–13.
Windebank AJ, Blexrud MD, de Groen PC. Potential neurotoxicity of the solvent vehicle for cyclosporine. J Pharmacol Exp Ther. 1994;268(2):1051–6.
Sanchez H, Bigard X, Veksler V, et al. Immunosuppressive treatment affects cardiac and skeletal muscle mitochondria by the toxic effect of vehicle. J Mol Cell Cardiol. 2000;32(2):323–31.
Bowers VD, Locker S, Ames S, et al. The hemodynamic effects of Cremophor-EL. Transplantation. 1991;51(4):847–50.
Theis JG, Liau-Chu M, Chan HS, et al. Anaphylactoid reactions in children receiving high-dose intravenous cyclosporine for reversal of tumor resistance: the causative role of improper dissolution of Cremophor EL. J Clin Oncol. 1995;13(10):2508–16.
Liau-Chu M, Theis JG, Koren G. Mechanism of anaphylactoid reactions: improper preparation of high-dose intravenous cyclosporine leads to bolus infusion of Cremophor EL and cyclosporine. Ann Pharmacother. 1997;31(11):1287–91.
Gotardo MA, Monteiro M. Migration of diethylhexyl phthalate from PVC bags into intravenous cyclosporine solutions. J Pharm Biomed Anal. 2005;38(4):709–13.
Venkataramanan R, Burckart GJ, Ptachcinski RJ, et al. Leaching of diethylhexyl phthalate from polyvinyl chloride bags into intravenous cyclosporine solution. Am J Hosp Pharm. 1986;43(11):2800–2.
Trapani G, Altomare C, Liso G, et al. Propofol in anesthesia. Mechanism of action, structure-activity relationships, and drug delivery. Curr Med Chem. 2000;7(2):249–71.
Mattila MA, Rossi ML, Ruoppi MK, et al. Reduction of venous sequelae of i.v. diazepam with a fat emulsion as solvent. Br J Anaesth. 1981;53(12):1265–8.
Micha JP, Goldstein BH, Birk CL, et al. Abraxane in the treatment of ovarian cancer: the absence of hypersensitivity reactions. Gynecol Oncol. 2006;100(2):437–8.
Morgan M, Whitwam JG. Althesin. Anaesthesia. 1985;40(2):121–3.
Guidance for Industry: Statistical Approaches to Establishing Bioequivalence. Rockville, MD, USA: United States Department of Health and Human Services, Food and Drug Administration (FDA); 2001.
Note for Guidance on the Investigation of Bioavailability and Bioequivalence. London, Great Britain: European Medicines Agency (EMA); 2001.
Guidance for Industry: Bioanalytical Method Validation. Rockville, MD, USA: United States Department of Health and Human Services, Food and Drug Administration (FDA); 2001.
Diletti E, Hauschke D, Steinijans VW. Sample size determination for bioequivalence assessment by means of confidence intervals. Int J Clin Pharmacol Ther Toxicol. 1992;30(Suppl 1):S51–8.
Kanto JH. Propofol, the newest induction agent of anesthesia. Int J Clin Pharmacol Ther Toxicol. 1988;26(1):41–57.
Kloover JS, den Bakker MA, Gelderblom H, et al. Fatal outcome of a hypersensitivity reaction to paclitaxel: a critical review of premedication regimens. Br J Cancer. 2004;90(2):304–5.
Cook AM, Whitlow J, Hatton J, et al. Cyclosporine A for neuroprotection: establishing dosing guidelines for safe and effective use. Expert Opin Drug Saf. 2009;8(4):411–9.
Gupta SK, Manfro RC, Tomlanovich SJ, et al. Effect of food on the pharmacokinetics of cyclosporine in healthy subjects following oral and intravenous administration. J Clin Pharmacol. 1990;30(7):643–53.
Christians U, Klawitter J, Clavijo CF. Bioequivalence testing of immunosuppressants: concepts and misconceptions. Kidney Int Suppl. 2010;115:S1–7.
Christians U, First MR, Benet LZ. Recommendations for bioequivalence testing of cyclosporine generics revisited. Ther Drug Monit. 2000;22(3):330–45.
Kahan BD, Dunn J, Fitts C, et al. Reduced inter- and intrasubject variability in cyclosporine pharmacokinetics in renal transplant recipients treated with a microemulsion formulation in conjunction with fasting, low-fat meals, or high-fat meals. Transplantation. 1995;59(4):505–11.
Kees F, Bucher M, Schweda F, et al. Neoimmun versus Neoral: a bioequivalence study in healthy volunteers and influence of a fat-rich meal on the bioavailability of Neoimmun. Naunyn Schmiedebergs Arch Pharmacol. 2007;375(6):393–9.
Christians U, Schmitz V, Haschke M. Functional interactions between P-glycoprotein and CYP3A in drug metabolism. Expert Opin Drug Metab Toxicol. 2005;1(4):641–54.
Kovarik JM, Mueller EA, van Bree JB, et al. Cyclosporine pharmacokinetics and variability from a microemulsion formulation—a multicenter investigation in kidney transplant patients. Transplantation. 1994;58(6):658–63.
Dutta S, Matsumoto Y, Ebling WF. Propofol pharmacokinetics and pharmacodynamics assessed from a Cremophor EL formulation. J Pharm Sci. 1997;86(8):967–9.
Fee JP, Dundee JW, Collier PS, et al. Bioavailability of intravenous diazepam. Lancet. 1984;2(8406):813.
Fee JP, Collier PS, Dundee JW. Bioavailability of three formulations of intravenous diazepam. Acta Anaesthesiol Scand. 1986;30(4):337–40.
Lee M, Min DI, Ku YM, et al. Effect of grapefruit juice on pharmacokinetics of microemulsion cyclosporine in African American subjects compared with Caucasian subjects: does ethnic difference matter? J Clin Pharmacol. 2001;41(3):317–23.
Min DI, Lee M, Ku YM, et al. Gender-dependent racial difference in disposition of cyclosporine among healthy African American and white volunteers. Clin Pharmacol Ther. 2000;68(5):478–86.
Acknowledgments
All authors receive salary support from, and are stockholders of, NeuroVive Pharmaceutical AB (publ). In addition, Eskil Elmér is co-founder and officer of Maas Biolab, LLC, and NeuroVive Pharmaceutical AB, which hold intellectual property rights and develop cyclophilin-D-inhibiting ciclosporins primarily for the treatment of acute brain injury and reperfusion injury in myocardial infarction. There are no other conflicts of interest to disclose. The authors are grateful to Marcus F. Keep, Daniel Wermeling and Torben Balchen for extensive input on the clinical trial protocol.
Author information
Authors and Affiliations
Corresponding author
Additional information
ClinicalTrials.gov identifier NCT01692834.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ehinger, K.H.J., Hansson, M.J., Sjövall, F. et al. Bioequivalence and Tolerability Assessment of a Novel Intravenous Ciclosporin Lipid Emulsion Compared to Branded Ciclosporin in Cremophor® EL. Clinical Drug Investigation 33, 25–34 (2013). https://doi.org/10.1007/s40261-012-0029-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-012-0029-x