
Abstract
Inflammation has been closely linked to various forms of cancer. Less is known about the role of inflammation in glioma, especially at the initiation stage. In this review, we first describe the unique features of the immune system in the brain. We then discuss the current understanding of the mechanisms by which glioma cells modulate the immune system, especially how bi-directional communications between immune cells and glioma cells create an immunosuppressed microenvironment that promotes tumor survival and growth. We also address the potential tumor-initiating roles of inflammation in glioma. Finally, we describe several immunotherapy approaches currently being developed to reverse these interactions and stimulate the immune system to eliminate glioma cells.
Similar content being viewed by others


Introduction
Inflammation is a natural immune response to an infection, tissue injury or malfunction. An acute inflammatory response begins when tissue-resident macrophages and mast cells detect an infection or damage. These cells secrete pro-inflammatory molecules that trigger a localized increase in blood flow and extravasation of plasma proteins and leukocytes to the affected tissue, leading to the typical swelling and redness. Once the immune response has neutralized the threat, pro-inflammatory molecules are replaced with anti-inflammatory molecules and the inflammation subsides. However, if the cause(s) of the initial inflammatory response are not resolved, an acute inflammation can transition to a chronic inflammation, lasting weeks, months, or even years. Whereas acute inflammation is beneficial, helping to eliminate infectious agents and promote tissue repair, chronic inflammation can have deleterious effects, such as tissue damage, autoimmune disease, or even cancer growth.
While immune surveillance blocks tumor formation, inflammation is known to promote tumorigenesis in many circumstances [1–4]. For example, cancer can be caused by inflammation triggered by infections (Hepatitis B/C-associated hepatocellular carcinoma, H. pylori-associated gastric cancer), autoimmune diseases (colitis-associated colorectal cancer) and even environmental irritants (asbestos-associated mesothelioma). Inflammation can promote tumorigenesis via increased genetic alterations, resulting from macrophage-secreted reactive oxygen and nitrogen species or activation-induced cytidine deaminase, a mutagenic enzyme. In the gut, inflammation strips away protective mucosal layers, exposing stem cells to environmental carcinogens and releasing them from normal homeostatic controls. Additionally, some cytokines secreted during the inflammatory response promote tumor growth by inducing angiogenesis, or by triggering signaling cascades that activate NFκB and STAT3, which in turn activate proliferative and anti-apoptotic genes [5, 6]. Three interesting themes emerge from these studies. First, inflammation can either inhibit or promote tumor growth, depending on the combination of immune cells present and the signaling factors they secrete. Second, the impact of inflammation on mutant cells could evolve as tumors progress from benign to malignant stages. Third, the interactions between immune and tumor cells are bi-directional, which adds another layer of complexity to this problem.
The role of inflammation in glioma is less clear than in the aforementioned cancer types. Gliomas are a form of cancer in the central nervous system (CNS) with diverse pathological and histological properties. The most common form, glioblastoma multiforme (GBM), is one of the deadliest of all cancers and has a median survival period of 12–14 months. Epidemiological studies have suggested a link between inflammation and glioma. Understanding how the immune system and gliomas interact could lead to novel therapeutic approaches to combat glioma. In this review, we will discuss the unique features of immunity in the CNS, the potential roles of inflammation in causing gliomagenesis, the known effects of inflammation in malignant gliomas, and how we could take advantage of the interactions between immune and tumor cells to more effectively treat glioma patients.
CNS Immune System
The brain is commonly known as an immune-privileged organ due to the restrictive nature of the blood–brain barrier (BBB) that prevents immune cells and serum-derived immune modulators from accessing it [7]. In the absence of pathological damage, the only cells with immune functions in the CNS parenchyma are microglia. However, immune cells from the hematopoietic system play important roles in the brain when the BBB is compromised by physical trauma or pathological conditions including multiple sclerosis, stroke, and malignant brain tumors.
Microglia
Microglia share a common origin with the hematopoietic system but segregate into a unique lineage early in development. They are generated from primitive myeloid progenitors in the extra-embryonic yolk sac before E8 [8]. After entering the CNS at E10 [8•, 9], microglia appear to replenish themselves solely through self-renewal without the involvement of peripheral macrophages, their bone marrow-derived close relatives.
In a healthy CNS, branched (resting) microglia actively monitor their environment by constantly extending/retracting their processes, while maintaining a static cell body [10, 11]. A resting microglia is activated when it detects pathogen-associated molecular patterns (PAMPs; ex. LPS) or damage-associated molecular patterns (DAMPs; ex. extracellular matrix molecule fragments) through their binding to pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD) proteins, and c-type lectin receptors. Serum proteins and dysfunctional neurons can also trigger microglial activation (see below). Activated microglia become amoeboid and can exist in different states representing different outcomes of the immune response. Similarly to macrophages, microglia can be classified under M1 (classic activation) and M2 (includes alternative activation and acquired deactivation) activation states [12•]. Classically activated microglia upregulate the expression of MHCII, thus increasing their antigen-presenting ability to activate T cells, secrete pro-inflammatory cytokines including TNFα, IL6 and IL1β, and upregulate nitric oxide synthase (NOS) to produce NO to destroy pathogens. Macrophage transition from classic to alternative activation is regulated by T cell-derived IL4 and IL13. Although their source in the CNS is unclear, IL4 and IL13 can also shift microglia-mediated immune responses, from pro-inflammatory and destructive to anti-inflammatory and tissue regenerating, by downregulating TNFα, IL1β and NOS2 and upregulating arginase 1, mannose receptor, FIZZ1, and Ym1 [13, 14]. However, it is currently challenging to determine whether these effects are truly microglia-specific or if they were observed in macrophages infiltrating the CNS [12, 15]. Acquired deactivation also shifts microglial responses to an immunosuppressive state, but is mediated by TGFβ and/or IL10. Additionally, acquired deactivation is characterized by microglial phagocytosis of apoptotic cells, which also acts as a signal to “deactivate” microglia [12].
Infiltrating Immune Cells Upon BBB Breakdown
Activated microglia secrete pro-inflammatory cytokines such as IL1β, which modulate the BBB to allow bone marrow-derived immune cells into the CNS [16], including CD8+ cytotoxic T cells and CD4+ helper T cells. The latter are subdivided into Th1, Th2, and Th3, which have different effects on an immune response [17]. Th1 cells promote a pro-inflammatory state by secreting cytokines such as IFNγ and IL12, activating macrophages and microglia to release toxic compounds such as NO, and activating cytotoxic CD8+ T cells. On the other hand, Th2 cells secrete anti-inflammatory cytokines such as IL4, IL10, and IL13, which counteract the effects of Th1 cells and promote B lymphocyte activation and antibody production. The main role of Th3 cells is to suppress the immune response by secreting TGFβ. However, Th1 cells can also produce anti-inflammatory cytokines, such as IL10 and TGFβ, later in an immune response to limit the length of the cytotoxic, tissue-damaging phase of this response. Another important type of CD4+ T cell is the regulatory T cell (Treg), which has mainly anti-inflammatory, immunosuppressive roles due to the production of high levels of IL10 and TGFβ as well as other, unknown mechanisms. These cells play an important role in shutting down an immune response once the threat to the host has been eliminated as well as in prevention of autoimmune disease [18].
Macrophages also enter the CNS upon BBB breakdown. However, due to similar marker expression and functions to microglia, and other experimental caveats, it has been very difficult to study the distinct contributions of infiltrated macrophages versus resident microglia to CNS immune activities [19], particularly in pathological cases. Nonetheless, significant differences have been identified between these cell types. Compared to macrophages, microglia have attenuated immune properties, such as poor antigen-presenting ability (reduced MHCII levels) and reduced phagocytic ability. Additionally, the BBB shields microglia from serum proteins including fibrinogen, a strong activator for both macrophages and microglia [20]. Lastly, healthy neurons signal to microglia through a variety of receptor/ligand complexes that keep them in a resting state [21]. Overall, the unique properties of microglia in the CNS are likely critical to avoid excessive damage in an organ with limited regenerative ability.
The Impact of Astrocytes on CNS Immunity
Though not part of the immune system, astrocytes also play a significant role in modulating immune responses in the CNS [22]. Astrocyte end-feet form the Glia limitans, the last barrier to immune cell entry into the CNS [23]. Upon trauma or in autoimmune diseases, reactive astrocytes form a barrier to the infiltration of the CNS by peripheral leukocytes [24]. Similar to microglia, astrocytes express various PRR receptors, such as TLR3, and are thus capable of responding to pathogens and tissue damage. Once activated, reactive astrocytes can secrete pro-inflammatory cytokines and chemokines that can activate and recruit immune cells to the affected CNS region (IL6, CCL2, and others). Finally, reactive astrocytes can secrete trophic factors such as CNTF and IGF1 that participate in the tissue repair phase of inflammation [22]. It has also been suggested that astrocytes can act as antigen-presenting cells (APCs), thus directly participating in T cell activation, though this remains a controversial issue [25].
Inflammation in Glioma
To understand the role of inflammation in gliomagenesis, we will discuss both the initiation and the malignant stages since inflammatory cells and molecules are involved in both stages, though in different ways. We will also discuss the complex nature of this topic, especially the anti- and pro-tumor activities of inflammation and the two-way interactions between immune and tumor cells.
Can Inflammation Cause Glioma?
Epidemiological studies suggest a link between inflammation and gliomagenesis: individuals with a history of NSAID use, asthma, allergies, or high IgE levels have decreased risk of developing glioma, whereas use of anti-histamines (lowers IgE levels, used to treat allergies and asthma) has the opposite association [26–29]. It has been hypothesized that allergic reactions and high IgE serum levels might lead to lower levels of Tregs, a cell type known to cause chronic, tumor-promoting inflammation [30]. Additionally, a number of studies have revealed associations between SNPs on immune-related genes and risk of glioma [30, 31], including the anti-inflammatory IL4, IL4R, IL10, and IL13, and the pro-inflammatory Cox2 and IL6. However, functional studies will be needed to verify such associations. It is interesting to speculate that, similar to how inflammatory bowel disease can cause colorectal cancer, autoimmune diseases affecting the CNS might also contribute to gliomagenesis. In fact, some multiple sclerosis patients develop glioma in the lesioned areas years later [32, 33]. A similar link has been suspected for head trauma [34, 35]. However, these associations are not highly significant due to the relatively low number of cases.
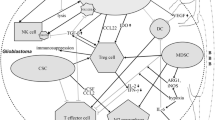
One intriguing possibility for how inflammation might influence gliomagenesis is by affecting the behavior of glioma cells of origin. Recent studies have identified both neural stem cells (NSCs) and oligodendrocyte precursors (OPCs) as cell types that, when mutated, can give rise to malignant gliomas [36•• , 37••, 38••]. Interestingly, molecules produced during inflammation can affect the behavior of both cell types (Fig. 1). Activated microglia and astrocytes secrete MCP1/CCL2 and SDF1 that can promote NSC migration to the inflamed area [39–42]. Pro-inflammatory cytokines can also recruit OPCs to promote repair of demyelinated lesions, and TNFα in particular can promote OPC and NSC proliferation [43–45]. Additionally, alternatively-activated microglia upregulate arginase 1, leading to the production of polyamines, which can promote NSC proliferation [46]. Conversely, TGFβ, which can be secreted by microglia, has well-known anti-proliferative effects on many types of CNS progenitors [47]. However, these effects are highly context-dependent, as microglia in the classic or alternative activation states can respectively inhibit or promote neurogenesis [48]. Similarly, high or low levels of IFNγ can inhibit or promote neurogenesis [49–51], and TNFα signaling through TNFR-I receptor inhibits hippocampal progenitor proliferation, whereas signaling through TNFR-II has the opposite effect, particularly in pathological conditions [52]. The study of interactions between neuroglial progenitors and immune cells is in its infancy, but has the potential to deliver critical insights into the effect of inflammation on glioma initiation.

Effects of inflammation on glioma initiation and growth. While the depicted cellular interactions are known to occur, a link between inflammation and glioma progression has only been suspected. Inflammatory molecules can affect immune cells (pro- or anti-inflammatory) and glioma cells of origin/glioma cells (anti- or pro-growth). Some inflammatory molecules can be pro- or anti-growth depending on the circumstances (see text for details; not all cells or interactions were represented)
Inflammation in the Malignant Phase: Friend or Foe?
The inflammatory response to a glioma is complex, with multiple cell types involved in multi-lateral communications (Fig. 1). Because gliomas cause a breakdown of the BBB, circulating immune cells not normally found in the CNS now gain access to tumor areas. This includes various types of T cells, B cells, macrophages, and myeloid-derived suppressor cells (MDSCs). Additionally, astrocytes become reactive and also participate in the response to the tumor. Finally, the tumor cells themselves secrete a number of factors that modulate the activity of all the cells mentioned above (as well as endothelial cells).
It has been estimated that up to 30 % of the glioma mass is composed of microglia [53]. Though these estimates can vary [54], the importance of microglia to the glioma microenvironment is unquestionable. Tumor-derived chemokines (MCP1/CCL2, MCP3, CSF1, G-CSF, HGF/SF) [55–61] attract microglia to gliomas, where they become activated. However, rather than engaging in cytotoxic or phagocytic activities, glioma-associated microglia tend to secrete cytokines and growth factors that promote tumor cell growth and angiogenesis, such as IL10 [62, 63], EGF [64], and VEGF [65]. Microglia-derived IL10 can also inhibit T cell proliferation and confer an immunosuppressed state. Glioma-associated microglia express MHCII and B7 molecules at the cell surface [66, 67]. This would allow microglia to act as APCs and activate a cytotoxic T cell response (via Th1 activation). However, glioma-associated microglia appear to have low APC activity [68], as they express lower levels of MHCII molecules than microglia activated in the absence of tumor cells [66, 67, 69] and lack expression of co-stimulatory molecules CD86, CD80, and CD40 [54]. Data suggest that this inhibition of microglial antigen-presenting capability is at least partially due to glioma-derived IL10 and prostaglandins [70]. In fact, many cytokines secreted by glioma cells can directly inhibit MHCII production in microglia [71]. Furthermore, glioma-associated microglia can actively eliminate T cells via FasL- and B7-H1-mediated apoptosis of CD8+ T cells [72, 73]. Decreased antigen presentation and T cell apoptosis are normal microglial negative feedback mechanisms to avoid destructive inflammation in the CNS, which are apparently hijacked by glioma cells to avoid elimination by the immune system.
In fact, the glioma microenvironment is strongly immunosuppressive, reminiscent of the M2 phase of inflammatory responses. Hao and colleagues [74] characterized the expression of 53 cytokines and receptors in multiple human tumors and found that the Th2/3 (immunosuppressive) profile was dominant over the Th1 (pro-inflammatory) profile. Consistent with this finding, TLR stimulation in glioma-associated microglia does not trigger classic activation, since these cells fail to secrete pro-inflammatory cytokines (IL6, IL1β, TNFα) and are thus incapable of activating a cytotoxic state in CD8+ T cells [54]. This immunosuppressive state is due to multiple factors secreted by glioma cells themselves. Glioma-derived M-CSF can drive glioma-associated microglia towards an M2 phenotype [75]. Other factors also increase prostaglandin-E2 (PGE2) production by microglia, which then has an autocrine effect decreasing microglial production of TNFα [76]. Glioma cells are known to express IL4 and PGE2 [77••], both of which can induce microglial alternative activation (see above). Similarly, TGFβ, a strong inducer of acquired deactivation [12], is also expressed by glioma cells [78, 79]. TGFβ plays a number of roles in the glioma microenvironment, including suppression of pro-inflammatory responses from T cells and microglia, but can also act directly on glioma cells to promote their growth. Several reports indicate that a subpopulation of glioma cells with tumor-initiating ability (cancer stem cells) are direct targets for TGFβ pro-tumorigenic effects [80–82]. Interestingly, TGFβ signaling has opposite effects in the pre-malignant and malignant phases of cancer for reasons that are just beginning to become apparent [47, 83]. In addition to anti-inflammatory cytokines, glioma cells also secrete IL6 and LIF [74, 84, 85], which could stimulate microglial classic activation or act on the glioma cells themselves to promote proliferation [86], and pro-angiogenic VEGF [65, 87], giving the tumor access to nutrients and oxygen. The glioma immunosuppressive environment is further enhanced by elevated numbers of Treg [54, 88]. These cells are recruited by glioma-derived chemoattractants (CCL2, CCL22) and can suppress T cell proliferation and promote an anti-inflammatory profile [89, 90]. The mechanisms by which Tregs exert these effects are still unclear, but it appears that expression of heme oxygenase 1 (HO1) plays an important role, perhaps via the production of carbon monoxide [91, 92]. Other mechanisms involve CTLA-4 and TGFβ [93]. Importantly, depletion of Tregs in mouse glioma models improves survival [94].
There are indications that, just as in other forms of cancer where inflammation plays a pro-tumorigenic role, STAT3 and NFκB, key nodes of cytokine intracellular signaling, are central to this effect. STAT3 inactivation changes the profile of glioma-associated microglia from anti- to pro-inflammatory and decreases glioma growth [95–97]; NFκB activity seems to block glioma cell differentiation while its inhibition promotes glioma cell senescence [98], whereas deletion of NFKBIA, an NFκB inhibitor, is associated with more aggressive gliomas [99].
In addition to immune cells, reactive astrocytes are also commonly found within gliomas. These cells secrete Shh [100–102], SDF1 [103–105], and IL-10 [62], which support glioma cell survival and proliferation. Astrocytes also play important roles for the enhancement of glioma cell migration, a property of gliomas that makes them particularly difficult to cure. UPA/uPAR from astrocytes cleaves plasminogen secreted by the glioma cells to plasmin, which in turn cleaves astrocyte-secreted pro-MMP2 to MMP2, thus increasing glioma cell invasiveness [106]. Additionally, glioma-secreted factors stimulate microglia to release MT1-MMP (MMP14) which can then activate glioma-derived pro-MMP2 [107]. Other molecular interactions and cellular players in the glioma microenvironment are not covered in this review due to space constraint, but we refer interested readers to recent reviews for more details [77••, 108••].
In summary, while the immune system can detect the presence of a glioma in the CNS and responds with mobilization/activation of many cell types, this response appears to be subverted from a pro-inflammatory, classic activation state to an immunosuppressed, alternative activation state by glioma-derived factors. If this tolerant, subverted state can be changed back to a pro-inflammatory state, then the immune system could help eliminate the glioma cells. Multiple efforts are currently underway to devise therapeutic strategies based on that concept.
Glioma Immunotherapy
The concept of cancer immunotherapy has existed for many years. It is based on recruiting the patient’s immune system and enhancing its anti-cancer properties to help eliminate the tumor cells [109•]. Passive immunotherapies use immune effector molecules (antibodies and cytokines) to elicit a rapid, but short-term, anti-tumor response. Active immunotherapies enhance T cell anti-tumor activity (autologous grafting of APCs exposed to tumor peptides or direct peptide injection) and can therefore confer a longer-lasting protection against cancer. Comprehensive reviews of the current state of glioma immunotherapy can be found in recent publications [110••, 111••]. Below, we discuss examples of promising glioma immunotherapies as well as approaches that have shown promise in other types of cancer and might soon be used against glioma.
One of the prerequisites of immunotherapy is the existence of antigens specific to glioma cells, a rare occurrence since tumors are composed of “self” cells. One candidate is EGFRvIII mutant receptor, which is not expressed by normal cells and has a prevalence of 20–30 % in GBM patients [112]. Immunization against EGFRvIII-specific antigens has shown safety and increased survival in glioma mouse models and patients [113, 114]. Unfortunately, immunotherapies based on EGFRvIII epitope alone eventually recur from tumor cells that had no EGFRvIII expression [114], illustrating the need for immunization against multiple tumor epitopes by using inactivated whole tumor cells as the immunogen [115–119]. Recently, several groups have reported high levels of cytomegalovirus (CMV) in human gliomas [120, 121], sparking interest in using “non-self” CMV antigens to mount an anti-glioma immune response [118].
In addition to unique antigens, immunotherapy can only be effective after overcoming the immunosuppressive environment created by gliomas. Some attempts have focused on inactivation of Tregs: CD25-blocking antibodies (IL2Rα, central to Treg function) have been used with some success in mice and humans [94, 122–124], and TLR8 and TLR9 stimulation can reduce Treg accumulation in glioma and improve CD8+ T cell activity [125, 126]. Other approaches could be to inhibit the CCL2/CCL22 chemokine receptor CCR4 to prevent Tregs from being attracted to the glioma site [89, 90].
Recent successes in immunotherapy for other cancers have re-ignited interest in this field [109]. Activity-blocking CTLA-4 antibodies are showing promise in the treatment of metastatic melanoma. This approach could also prove effective against glioma, where CTLA-4 plays a role in Treg-mediated immunosuppression. CTLA-4 also plays a direct role in downregulating effector T cell activity, and thus inhibiting it in gliomas could also improve these cells’ function in the tumor, provided there are cells with antigen-presenting ability to stimulate them. Another attractive candidate target for immunotherapy is PD-1, the receptor for PD-L1 (B7-H1), which can reduce proliferation and induce apoptosis of CD8+ T cells. PD-1-inactivating antibodies have been tested with encouraging results in phase I clinical trials for metastatic melanoma, colorectal cancer, non-small cell lung cancer, prostate cancer, and renal cell carcinoma [127]. As the mechanisms mediated by PD-1 and its ligand are known to occur in glioma (see above), this approach could also one day be beneficial for glioma patients.
Conclusion
Just as many parasites have developed strategies to evade immune rejection [128–130], gliomas seem to use similar approaches to avoid elimination by the immune system. By secreting immune-modulating factors, glioma cells convert microglial and macrophage activation away from a pro-inflammatory, classic state towards an anti-inflammatory, alternative activation state. This permissive environment is further enhanced by attracting Treg and other immunosuppressive cells into the tumor mass. Such modulations not only passively allow tumor cells to survive but can also promote their growth, possibly by engaging immune cells into a tissue repair mode. Therefore, rigorous studies are urgently needed to uncover the two-way communications between glioma and immune cells. Importantly, to fully understand the impact of inflammation on gliomagenesis, we should study such relationships not only in malignant gliomas but also during tumor-initiation stages, where many important questions remain to be answered. Do pre-malignant mutant cells trigger microglial activation, and if yes, then by which mechanism? If no, how do they evade detection? Do microglia initially try to eliminate mutant cells, or can mutant cells subvert microglial responses from the earliest stages towards a growth/survival-promoting role? Do these interactions accelerate gliomagenesis? Genetic mouse models of glioma can be particularly helpful in answering these questions due to the ability to follow mutant cells throughout their malignant progression in a physiologically relevant environment. Ultimately, therapeutic strategies that can thwart the subversive tricks of tumor cells and turn the immune system against them will yield tremendous clinical benefits. The challenges are great, but collaborations between immunologists, neuroscientists, and oncologists should make a cure possible.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Balkwill FR, Mantovani A (2012) Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol 22(1):33–40. doi:10.1016/j.semcancer.2011.12.005
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140(6):883–899. doi:10.1016/j.cell.2010.01.025
Grivennikov SI, Karin M (2010) Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev 20(1):65–71. doi:10.1016/j.gde.2009.11.004
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454(7203):436–444. doi:10.1038/nature07205
DiDonato JA, Mercurio F, Karin M (2012) NF-kappaB and the link between inflammation and cancer. Immunol Rev 246(1):379–400. doi:10.1111/j.1600-065X.2012.01099.x
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9(11):798–809. doi:10.1038/nrc2734
Wilson EH, Weninger W, Hunter CA (2010) Trafficking of immune cells in the central nervous system. J Clin Investig 120(5):1368–1379. doi:10.1172/JCI41911
• Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330(6005):841–845. doi:10.1126/science.1194637.
This paper provides an important contribution to the disputed question regarding the origin of adult microglia, convincingly showing that they arise early on in development and do not receive input from postnatal hematopoietic progenitors.
Alliot F, Lecain E, Grima B, Pessac B (1991) Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc Natl Acad Sci USA 88(4):1541–1545
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8(6):752–758. doi:10.1038/nn1472
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. doi:10.1126/science.1110647
• Colton CA (2009) Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 4(4):399–418. doi:10.1007/s11481-009-9164-4.
A good review of the different activation states of microglia and macrophages.
Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP (2006) Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflamm 3:27. doi:10.1186/1742-2094-3-27
Lyons A, Griffin RJ, Costelloe CE, Clarke RM, Lynch MA (2007) IL-4 attenuates the neuroinflammation induced by amyloid-beta in vivo and in vitro. J Neurochem 101(3):771–781. doi:10.1111/j.1471-4159.2006.04370.x
Olah M, Biber K, Vinet J, Boddeke HW (2011) Microglia phenotype diversity. CNS Neurol Disord Drug Targets 10(1):108–118
Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O’Banion MK (2007) Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosc 27(35):9301–9309. doi:10.1523/JNEUROSCI.1418-07.2007
Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ (2012) Helper T cell diversity and plasticity. Curr Opin Immunol 24(3):297–302. doi:10.1016/j.coi.2012.01.014
Sakaguchi S (2012) Naturally arising Foxp3-expressing CD25+CD4+regulatory T cells in immunological tolerance to self and non-self. Nat Immunol 6(4):345–352. doi:10.1038/ni1178
Ransohoff RM, Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145. doi:10.1146/annurev.immunol.021908.132528
Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H et al (2007) The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med 204(3):571–582. doi:10.1084/jem.20061931
Biber K, Neumann H, Inoue K, Boddeke HW (2007) Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci 30(11):596–602. doi:10.1016/j.tins.2007.08.007
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28(3):138–145. doi:10.1016/j.it.2007.01.005
Owens T, Bechmann I, Engelhardt B (2008) Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 67(12):1113–1121. doi:10.1097/NEN.0b013e31818f9ca8
Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S et al (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29(37):11511–11522. doi:10.1523/JNEUROSCI.1514-09.2009
De Keyser J, Laureys G, Demol F, Wilczak N, Mostert J, Clinckers R (2010) Astrocytes as potential targets to suppress inflammatory demyelinating lesions in multiple sclerosis. Neurochem Int 57(4):446–450. doi:10.1016/j.neuint.2010.02.012
Linos E, Raine T, Alonso A, Michaud D (2007) Atopy and risk of brain tumors: a meta-analysis. J Natl Cancer Inst 99(20):1544–1550. doi:10.1093/jnci/djm170
Scheurer ME, Amirian ES, Davlin SL, Rice T, Wrensch M, Bondy ML (2011) Effects of antihistamine and anti-inflammatory medication use on risk of specific glioma histologies. Int J Cancer 129(9):2290–2296. doi:10.1002/ijc.25883
Scheurer ME, El-Zein R, Thompson PA, Aldape KD, Levin VA, Gilbert MR et al (2008) Long-term anti-inflammatory and antihistamine medication use and adult glioma risk. Cancer Epidemiol Biomark Prev 17(5):1277–1281. doi:10.1158/1055-9965.EPI-07-2621
Sivak-Sears NR, Schwartzbaum JA, Miike R, Moghadassi M, Wrensch M (2004) Case-control study of use of nonsteroidal antiinflammatory drugs and glioblastoma multiforme. Am J Epidemiol 159(12):1131–1139. doi:10.1093/aje/kwh153
Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il’yasova D et al (2008) Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer 113(7 Suppl):1953–1968. doi:10.1002/cncr.23741
Gu J, Liu Y, Kyritsis AP, Bondy ML (2009) Molecular epidemiology of primary brain tumors. Neurotherapeutics 6(3):427–435. doi:10.1016/j.nurt.2009.05.001
de la Lama A, Gomez PA, Boto GR, Lagares A, Ricoy JR, Alen JF et al (2004) Oligodendroglioma and multiple sclerosis. A case report. Neurocirugia 15(4):378–383
Green AJ, Bollen AW, Berger MS, Oksenberg JR, Hauser SL (2001) Multiple sclerosis and oligodendroglioma. Mult Scler 7(4):269–273
Morantz RA, Shain W (1978) Trauma and brain tumors: an experimental study. Neurosurgery 3(2):181–186
Salvati M, Caroli E, Rocchi G, Frati A, Brogna C, Orlando ER (2004) Post-traumatic glioma. Report of four cases and review of the literature. Tumori 90(4):416–419
•• Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK et al. (2009) Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer cell 15(1):45–56. doi:10.1016/j.ccr.2008.12.006.
This paper identifies adult neural stem cells as a cell of origin for glioma in vivo.
•• Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H et al. (2011) Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146(2):209–221. doi:10.1016/j.cell.2011.06.014.
This paper identifies oligodendrocyte precursor cells as a cell of origin for glioma in vivo and that, even when mutations are targeted to neural stem cells, malignant transformation only occurs in the oligodendrocyte lineage, thus distinguishing the concepts of cell of mutation and cell of origin for glioma.
•• Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R et al. (2010) Non-stem cell origin for oligodendroglioma. Cancer Cell 18(6):669–682. doi:10.1016/j.ccr.2010.10.033.
This paper studies the origins of oligodendroglioma, a subtype of glioma, and provides evidence suggesting that these tumors originate from oligodendrocyte precursor cells.
Aarum J, Sandberg K, Haeberlein SL, Persson MA (2003) Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci USA 100(26):15983–15988. doi:10.1073/pnas.2237050100
D’Mello C, Le T, Swain MG (2009) Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29(7):2089–2102. doi:10.1523/JNEUROSCI.3567-08.2009
Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD et al (2004) Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA 101(52):18117–18122. doi:10.1073/pnas.0408258102
Widera D, Holtkamp W, Entschladen F, Niggemann B, Zanker K, Kaltschmidt B et al (2004) MCP-1 induces migration of adult neural stem cells. Eur J Cell Biol 83(8):381–387
Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP (2001) TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 4(11):1116–1122. doi:10.1038/nn738
Rubio-Araiz A, Arevalo-Martin A, Gomez-Torres O, Navarro-Galve B, Garcia-Ovejero D, Suetterlin P et al (2008) The endocannabinoid system modulates a transient TNF pathway that induces neural stem cell proliferation. Mol Cell Neurosci 38(3):374–380. doi:10.1016/j.mcn.2008.03.010
Widera D, Mikenberg I, Elvers M, Kaltschmidt C, Kaltschmidt B (2006) Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci 7:64. doi:10.1186/1471-2202-7-64
Malaterre J, Strambi C, Aouane A, Strambi A, Rougon G, Cayre M (2004) A novel role for polyamines in adult neurogenesis in rodent brain. Eur J Neurosci 20(2):317–330. doi:10.1111/j.1460-9568.2004.03498.x
Aigner L, Bogdahn U (2008) TGF-beta in neural stem cells and in tumors of the central nervous system. Cell Tissue Res 331(1):225–241. doi:10.1007/s00441-007-0466-7
Ekdahl CT, Kokaia Z, Lindvall O (2009) Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158(3):1021–1029. doi:10.1016/j.neuroscience.2008.06.052
Baron R, Nemirovsky A, Harpaz I, Cohen H, Owens T, Monsonego A (2008) IFN-gamma enhances neurogenesis in wild-type mice and in a mouse model of Alzheimer’s disease. FASEB J 22(8):2843–2852. doi:10.1096/fj.08-105866
Butovsky O, Landa G, Kunis G, Ziv Y, Avidan H, Greenberg N et al (2006) Induction and blockage of oligodendrogenesis by differently activated microglia in an animal model of multiple sclerosis. J Clin Invest 116(4):905–915. doi:10.1172/JCI26836
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S et al (2006) Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci 31(1):149–160. doi:10.1016/j.mcn.2005.10.006
Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z et al (2006) Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci 26(38):9703–9712. doi:10.1523/JNEUROSCI.2723-06.2006
Giometto B, Bozza F, Faresin F, Alessio L, Mingrino S, Tavolato B (1996) Immune infiltrates and cytokines in gliomas. Acta Neurochir 138(1):50–56
Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB (2006) The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-oncology 8(3):261–279. doi:10.1215/15228517-2006-008
Alterman RL, Stanley ER (1994) Colony stimulating factor-1 expression in human glioma. Mol Chem Neuropathol 21(2–3):177–188. doi:10.1007/BF02815350
Badie B, Schartner J, Klaver J, Vorpahl J (1999) In vitro modulation of microglia motility by glioma cells is mediated by hepatocyte growth factor/scatter factor. Neurosurgery 44(5):1077–1082 discussion 82–83
Leung SY, Wong MP, Chung LP, Chan AS, Yuen ST (1997) Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathol 93(5):518–527
Mueller MM, Herold-Mende CC, Riede D, Lange M, Steiner HH, Fusenig NE (1999) Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol 155(5):1557–1567. doi:10.1016/S0002-9440(10)65472-7
Nitta T, Sato K, Allegretta M, Brocke S, Lim M, Mitchell DJ et al (1992) Expression of granulocyte colony stimulating factor and granulocyte-macrophage colony stimulating factor genes in human astrocytoma cell lines and in glioma specimens. Brain Res 571(1):19–25
Okada M, Saio M, Kito Y, Ohe N, Yano H, Yoshimura S et al (2009) Tumor-associated macrophage/microglia infiltration in human gliomas is correlated with MCP-3, but not MCP-1. Int J Oncol 34(6):1621–1627
Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S et al (2003) Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann Neurol 54(3):388–392. doi:10.1002/ana.10679
Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC (1997) Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res 17(5A):3217–3224
Wagner S, Czub S, Greif M, Vince GH, Suss N, Kerkau S et al (1999) Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int J Cancer 82(1):12–16
Briers TW, Desmaretz C, Vanmechelen E (1994) Generation and characterization of mouse microglial cell lines. J Neuroimmunol 52(2):153–164
Lafuente JV, Adan B, Alkiza K, Garibi JM, Rossi M, Cruz-Sanchez FF (1999) Expression of vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor-beta (PDGFR-beta) in human gliomas. J Mol Neurosci 13(1–2):177–185. doi:10.1385/JMN:13:1-2:177
Badie B, Bartley B, Schartner J (2002) Differential expression of MHC class II and B7 costimulatory molecules by microglia in rodent gliomas. J Neuroimmunol 133(1–2):39–45
Tran CT, Wolz P, Egensperger R, Kosel S, Imai Y, Bise K et al (1998) Differential expression of MHC class II molecules by microglia and neoplastic astroglia: relevance for the escape of astrocytoma cells from immune surveillance. Neuropathol Appl Neurobiol 24(4):293–301
Flugel A, Labeur MS, Grasbon-Frodl EM, Kreutzberg GW, Graeber MB (1999) Microglia only weakly present glioma antigen to cytotoxic T cells. Int J Dev Neurosci 17(5–6):547–556
Schartner JM, Hagar AR, Van Handel M, Zhang L, Nadkarni N, Badie B (2005) Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 51(4):279–285. doi:10.1002/glia.20201
Taniguchi Y, Ono K, Yoshida S, Tanaka R (2000) Antigen-presenting capability of glial cells under glioma-harboring conditions and the effect of glioma-derived factors on antigen presentation. J Neuroimmunol 111(1–2):177–185
O’Keefe GM, Nguyen VT, Benveniste EN (1999) Class II transactivator and class II MHC gene expression in microglia: modulation by the cytokines TGF-beta, IL-4, IL-13 and IL-10. Eur J Immunol 29(4):1275–1285. doi:10.1002/(SICI)1521-4141(199904)29:04<1275:AID-IMMU1275>3.0.CO;2-T
Badie B, Schartner J, Prabakaran S, Paul J, Vorpahl J (2001) Expression of Fas ligand by microglia: possible role in glioma immune evasion. J Neuroimmunol 120(1–2):19–24
Magnus T, Schreiner B, Korn T, Jack C, Guo H, Antel J et al (2005) Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J Neurosci 25(10):2537–2546. doi:10.1523/JNEUROSCI.4794-04.2005
Hao C, Parney IF, Roa WH, Turner J, Petruk KC, Ramsay DA (2002) Cytokine and cytokine receptor mRNA expression in human glioblastomas: evidence of Th1, Th2 and Th3 cytokine dysregulation. Acta Neuropathol 103(2):171–178. doi:10.1007/s004010100448
Komohara Y, Ohnishi K, Kuratsu J, Takeya M (2008) Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 216(1):15–24. doi:10.1002/path.2370
Nakano Y, Kuroda E, Kito T, Uematsu S, Akira S, Yokota A et al (2008) Induction of prostaglandin E2 synthesis and microsomal prostaglandin E synthase-1 expression in murine microglia by glioma-derived soluble factors. Laboratory investigation. J Neurosurg 108(2):311–319. doi:10.3171/JNS/2008/108/2/0311
•• Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H (2012) The brain tumor microenvironment. Glia 60(3):502–514.
An excellent review of the many cellular interactions that shape the glioma microenvironment; it covers aspects that could not be mentioned in our review, including the tumor vascular niche and endothelial-glioma interactions.
Bodmer S, Strommer K, Frei K, Siepl C, de Tribolet N, Heid I et al (1989) Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J Immunol 143(10):3222–3229
Tada T, Yabu K, Kobayashi S (1993) Detection of active form of transforming growth factor-beta in cerebrospinal fluid of patients with glioma. Jpn J Cancer Res 84(5):544–548
Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA et al (2010) TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 18(6):655–668. doi:10.1016/j.ccr.2010.10.023
Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K (2009) Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 5(5):504–514. doi:10.1016/j.stem.2009.08.018
Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I et al (2009) TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 15(4):315–327. doi:10.1016/j.ccr.2009.02.011
Eyler CE, Rich JN (2012) Looking in the miR-ror: TGF-beta-mediated activation of NF-kappaB in glioma. J Clin Invest 122(10):3473–3475. doi:10.1172/JCI66058
Halfter H, Kremerskothen J, Weber J, Hacker-Klom U, Barnekow A, Ringelstein EB et al (1998) Growth inhibition of newly established human glioma cell lines by leukemia inhibitory factor. J Neurooncol 39(1):1–18
Van Meir E, Sawamura Y, Diserens AC, Hamou MF, de Tribolet N (1990) Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res 50(20):6683–6688
Goswami S, Gupta A, Sharma SK (1998) Interleukin-6-mediated autocrine growth promotion in human glioblastoma multiforme cell line U87MG. J Neurochem 71(5):1837–1845
Tsai JC, Goldman CK, Gillespie GY (1995) Vascular endothelial growth factor in human glioma cell lines: induced secretion by EGF, PDGF-BB, and bFGF. J Neurosurg 82(5):864–873. doi:10.3171/jns.1995.82.5.0864
El Andaloussi A, Lesniak MS (2006) An increase in CD4+CD25+FOXP3+regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro-oncology 8(3):234–243. doi:10.1215/15228517-2006-006
Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJ, Wesseling P et al (2010) Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol 225(1–2):195–199. doi:10.1016/j.jneuroim.2010.05.020
Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB (2008) Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother 57(1):123–131. doi:10.1007/s00262-007-0336-x
Pae HO, Oh GS, Choi BM, Chae SC, Kim YM, Chung KR et al (2004) Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J Immunol 172(8):4744–4751
Song R, Zhou Z, Kim PK, Shapiro RA, Liu F, Ferran C et al (2004) Carbon monoxide promotes Fas/CD95-induced apoptosis in Jurkat cells. J Biol Chem 279(43):44327–44334. doi:10.1074/jbc.M406105200
Sonabend AM, Rolle CE, Lesniak (2008) The role of regulatory T cells in malignant glioma. Anticancer Res 28(2B):1143–1150
El Andaloussi A, Han Y, Lesniak MS (2006) Prolongation of survival following depletion of CD4+CD25+regulatory T cells in mice with experimental brain tumors. J Neurosurg 105(3):430–437. doi:10.3171/jns.2006.105.3.430
Gao L, Li F, Dong B, Zhang J, Rao Y, Cong Y et al (2010) Inhibition of STAT3 and ErbB2 suppresses tumor growth, enhances radiosensitivity, and induces mitochondria-dependent apoptosis in glioma cells. Int J Radiat Oncol Biol Phys 77(4):1223–1231. doi:10.1016/j.ijrobp.2009.12.036
Michaud-Levesque J, Bousquet-Gagnon N, Beliveau R (2012) Quercetin abrogates IL-6/STAT3 signaling and inhibits glioblastoma cell line growth and migration. Exp Cell Res 318(8):925–935. doi:10.1016/j.yexcr.2012.02.017
Zhang L, Alizadeh D, Van Handel M, Kortylewski M, Yu H, Badie B (2009) Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia 57(13):1458–1467. doi:10.1002/glia.20863
Nogueira L, Ruiz-Ontanon P, Vazquez-Barquero A, Lafarga M, Berciano MT, Aldaz B et al (2011) Blockade of the NFkappaB pathway drives differentiating glioblastoma-initiating cells into senescence both in vitro and in vivo. Oncogene 30(32):3537–3548. doi:10.1038/onc.2011.74
Bredel M, Scholtens DM, Yadav AK, Alvarez AA, Renfrow JJ, Chandler JP et al (2011) NFKBIA deletion in glioblastomas. N Engl J Med 364(7):627–637. doi:10.1056/NEJMoa1006312
Becher OJ, Hambardzumyan D, Fomchenko EI, Momota H, Mainwaring L, Bleau AM et al (2008) Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res 68(7):2241–2249. doi:10.1158/0008-5472.CAN-07-6350
Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 17(2):165–172. doi:10.1016/j.cub.2006.11.033
Dahmane N, Sanchez P, Gitton Y, Palma V, Sun T, Beyna M et al (2001) The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development 128(24):5201–5212
Bajetto A, Bonavia R, Barbero S, Piccioli P, Costa A, Florio T et al (1999) Glial and neuronal cells express functional chemokine receptor CXCR4 and its natural ligand stromal cell-derived factor 1. J Neurochem 73(6):2348–2357
Rempel SA, Dudas S, Ge S, Gutierrez JA (2000) Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res 6(1):102–111
Zhou Y, Larsen PH, Hao C, Yong VW (2002) CXCR4 is a major chemokine receptor on glioma cells and mediates their survival. J Biol Chem 277(51):49481–49487. doi:10.1074/jbc.M206222200
Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG et al (2003) Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci 23(10):4034–4043
Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K et al (2009) Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci USA 106(30):12530–12535. doi:10.1073/pnas.0804273106
•• Rolle CE, Sengupta S, Lesniak MS (2012) Mechanisms of immune evasion by gliomas. Adv Exp Med Biol 746:53–76. doi:10.1007/978-1-4614-3146-6_5.
An excellent review on the multiple mechanisms used by glioma cells to evade the immune system; it covers aspects that could not be mentioned in our review such as the role of myeloid-derived suppressor cells in glioma immunosuppression.
• Mellman I, Coukos G, Dranoff G (2011) Cancer immunotherapy comes of age. Nature 480(7378):480–489. doi:10.1038/nature10673.
An excellent review of past and recent progress in the general field of cancer immunotherapy.
•• Johnson LA, Sampson JH (2010) Immunotherapy approaches for malignant glioma from 2007 to 2009. Curr Neurol Neurosci Rep 10(4):259–266. doi:10.1007/s11910-010-0111-9.
An excellent summary of success stories and failures in glioma immunotherapy research.
•• Okada H, Kohanbash G, Zhu X, Kastenhuber ER, Hoji A, Ueda R et al. (2009) Immunotherapeutic approaches for glioma. Crit Rev Immunol 29(1):1–42.
A very comprehensive review of different immunotherapy methods that have been tested specifically against glioma.
Gan HK, Kaye AH, Luwor RB (2009) The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci 16(6):748–754. doi:10.1016/j.jocn.2008.12.005
Heimberger AB, Archer GE, Crotty LE, McLendon RE, Friedman AH, Friedman HS et al (2002) Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery 50(1):158–164 discussion 64–66
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS et al (2010) Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 28(31):4722–4729. doi:10.1200/JCO.2010.28.6963
De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J et al (2008) Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res 14(10):3098–3104. doi:10.1158/1078-0432.CCR-07-4875
Ishikawa E, Tsuboi K, Yamamoto T, Muroi A, Takano S, Enomoto T et al (2007) Clinical trial of autologous formalin-fixed tumor vaccine for glioblastoma multiforme patients. Cancer Sci 98(8):1226–1233. doi:10.1111/j.1349-7006.2007.00518.x
Okada H, Lieberman FS, Walter KA, Lunsford LD, Kondziolka DS, Bejjani GK et al (2007) Autologous glioma cell vaccine admixed with interleukin-4 gene transfected fibroblasts in the treatment of patients with malignant gliomas. J Transl Med 5:67. doi:10.1186/1479-5876-5-67
Prins RM, Cloughesy TF, Liau LM (2008) Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Eng J Med 359(5):539–541. doi:10.1056/NEJMc0804818
Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S et al (2008) Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res 68(14):5955–5964. doi:10.1158/0008-5472.CAN-07-5973
Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH et al (2002) Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62(12):3347–3350
Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE et al (2008) Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro-oncology 10(1):10–18. doi:10.1215/15228517-2007-035
Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA et al (2006) Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res 12(14 Pt 1):4294–4305. doi:10.1158/1078-0432.CCR-06-0053
Grauer OM, Sutmuller RP, van Maren W, Jacobs JF, Bennink E, Toonen LW et al (2008) Elimination of regulatory T cells is essential for an effective vaccination with tumor lysate-pulsed dendritic cells in a murine glioma model. Int J Cancer 122(8):1794–1802. doi:10.1002/ijc.23284
Mitchell DA, Cui X, Schmittling RJ, Sanchez-Perez L, Snyder DJ, Congdon KL et al (2011) Monoclonal antibody blockade of IL-2 receptor alpha during lymphopenia selectively depletes regulatory T cells in mice and humans. Blood 118(11):3003–3012. doi:10.1182/blood-2011-02-334565
El Andaloussi A, Sonabend AM, Han Y, Lesniak MS (2006) Stimulation of TLR9 with CpG ODN enhances apoptosis of glioma and prolongs the survival of mice with experimental brain tumors. Glia 54(6):526–535. doi:10.1002/glia.20401
Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T et al (2005) Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 309(5739):1380–1384. doi:10.1126/science.1113401
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH et al (2010) Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 28(19):3167–3175. doi:10.1200/JCO.2009.26.7609
Maizels RM, Smith KA (2011) Regulatory T cells in infection. Adv Immunol 112:73–136. doi:10.1016/B978-0-12-387827-4.00003-6
Raes G, Beschin A, Ghassabeh GH, De Baetselier P (2007) Alternatively activated macrophages in protozoan infections. Curr Opin Immunol 19(4):454–459. doi:10.1016/j.coi.2007.05.007
Van Ginderachter JA, Beschin A, De Baetselier P, Raes G (2010) Myeloid-derived suppressor cells in parasitic infections. Eur J Immunol 40(11):2976–2985. doi:10.1002/eji.201040911
Acknowledgments
We thank Dr. Bin Zhang for critical comments on this manuscript. This work was supported in part by grants from the National Institutes of Health (R01-CA136495) and W.M. Keck Foundation.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Galvão, R.P., Zong, H. Inflammation and Gliomagenesis: Bi-Directional Communication at Early and Late Stages of Tumor Progression. Curr Pathobiol Rep 1, 19–28 (2013). https://doi.org/10.1007/s40139-012-0006-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-012-0006-3