
Abstract
Purpose
The purpose of this review is to summarize current knowledge of detailed biochemical evidence for the role of γ-aminobutyric acid type A receptors (GABAA–Rs) in the mechanisms of general anesthesia.
Principal findings
With the knowledge that all general anesthetics positively modulate GABAA-R-mediated inhibitory transmission, site-directed mutagenesis comparing sequences of GABAA-R subunits of varying sensitivity led to identification of amino acid residues in the transmembrane domain that are critical for the drug actions in vitro. Using a photo incorporable analogue of the general anesthetic, R(+)etomidate, we identified two transmembrane amino acids that were affinity labelled in purified bovine brain GABAA-R. Homology protein structural modelling positions these two residues, αM1-11’ and βM3-4’, close to each other in a single type of intersubunit etomidate binding pocket at the β/α interface. This position would be appropriate for modulation of agonist channel gating. Overall, available information suggests that these two etomidate binding residues are allosterically coupled to sites of action of steroids, barbiturates, volatile agents, and propofol, but not alcohols. Residue α/βM2-15’ is probably not a binding site but allosterically coupled to action of volatile agents, alcohols, and intravenous agents, and α/βM1-(-2’) is coupled to action of intravenous agents.
Conclusions
Establishment of a coherent and consistent structural model of the GABAA-R lends support to the conclusion that general anesthetics can modulate function by binding to appropriate domains on the protein. Genetic engineering of mice with mutation in some of these GABAA-R residues are insensitive to general anesthetics in vivo, suggesting that further analysis of these domains could lead to development of more potent and specific drugs.
Résumé
Objectif
L’objectif de cet article de synthèse est de résumer les connaissances actuelles concernant les données probantes biochimiques détaillées élucidant le rôle des récepteurs à l’acide γ-aminobutyrique de type A (R-GABAA) dans les mécanismes de l’anesthésie générale.
Constatations principales
Tous les anesthésiques généraux modulent positivement la transmission inhibitrice médiée par les R-GABAA. On a identifié les acides aminés du domaine transmembranaire représentant des sites d’action importants des médicaments, en effectuant des mutations ciblées sur des séquences de sous-unités des R-GABAA et en comparant leur sensibilité. À l’aide d’un analogue photo luminescent de l’anesthésique général étomidate R(+), nous avons identifié deux acides aminés transmembranaires marquées par affinité dans les R-GABAA purifiés de cerveau bovin. Un modèle structurel de protéine par homologie place ces deux résidus, soit αM1-11’ et βM3-4’, à proximité l’un de l’autre dans un type unique de poche de liaison d’étomidate inter-sous-unité au niveau de l’interface β/α. Cette position sera adaptée pour moduler le portillon des canaux agonistes. Globalement, les données disponibles suggèrent que des deux résidus se liant à l’étomidate sont couplés de façon allostérique aux sites d’action des stéroïdes, des barbituriques, des agents volatils et du propofol, mais pas à ceux des alcools. Le résidu α/βM2-15’ n’est probablement pas un site de liaison, mais il est couplé de façon allostérique à l’action des agents volatils, des alcools et des agents intraveineux, et le α/βM1-(-2’) est couplé à l’action des agents intraveineux.
Conclusion
La création d’un modèle structurel cohérent et logique des R-GABAA appuie notre conclusion selon laquelle les anesthésiques généraux peuvent moduler leur fonction en se liant à des domaines spécifiques sur la protéine. Les souris génétiquement modifiées porteuses d’une mutation dans certains de ces résidus de R-GABAA ne sont pas sensibles aux anesthésiques généraux in vivo, ce qui suggère qu’une analyse plus approfondie de ces domaines pourrait permettre la mise au point de médicaments à la fois plus puissants et plus spécifiques.
Similar content being viewed by others

The purpose of this review is to inform the reader of the current knowledge of detailed biochemical evidence for the role of γ-aminobutyric acid type A receptors (GABAA–Rs) in the mechanisms of anesthesia. We briefly summarize the evidence connecting anesthetic action on the brain with action on GABAA-Rs and the structure and function of GABAA-Rs. We describe the major techniques utilized for determination of amino acids and sequence domains within proteins that participate in various functions, and we summarize the newest data developed on the study of GABAA-Rs to identify such sites. We also discuss how the overall data for all the chemical classes of general anesthetics either form or do not form a coherent story for a role for GABAA-Rs in anesthetic action and its relevance for the future of clinical anesthesiology.
Most drugs that act as general anesthetics act only at relatively high concentrations, and their interaction with biosystems has long been considered as more of a physical interaction, namely, perturbation of fluidity of the membrane lipid bilayer, which is consistent with the Meyer-Overton correlation. In this correlation, potency as a general anesthetic is directly related to the solubility ratio for the compound in an oil-water mixture rather than to chemically specific binding to recognition sites on the molecules involved, as in traditional drug receptors.1 However, recent electrophysiological studies on specificity of action of anesthetic agents on biosystems and biochemical investigations on specific interactions with proteins, especially membrane channels, suggest that general anesthetics probably act via hydrophobic regions of protein targets in neurons.2,3 A major candidate for the target of general anesthetics is the GABAA receptor, a ligand-gated chloride ion channel receptor for the neurotransmitter GABA, which universally inhibits neuronal excitability via synaptic phasic currents as well as extrasynaptic tonic currents.4-7 The GABAA -Rs are actually a family of heteropentamers with differing subunit composition that vary in their age-dependence and brain and subcellular localization of expression and trafficking, as well as their pharmacological specificity, including sensitivity to GABA agonists and allosteric modulators, such as benzodiazepines, general anesthetics, neurosteroids, and ethanol (EtOH).8,9
General anesthetic action and GABAA-Rs
Most if not all general anesthetics at relevant concentrations enhance the function of GABAA-Rs, and GABAA-Rs are positioned in the nervous system where they can mediate anesthesia.3,7,10-12 People have used the differential sensitivity of GABAA-R subunits to anesthetic modulation, especially the low sensitivity of ρ subtypes, to attempt to identify amino acid residues in the GABAA-R subunit sequences that are critical for anesthetic modulation.13,14 The residues implicated are in or near the transmembrane domains. Virtually all of the chemical classes of adrugs with general anesthetic activity are implicated, including especially all of the intravenously administered agents, such as, etomidate, propofol, barbiturates, steroids, and alcohols, as well as the volatile gaseous agents. Some of these transmembrane GABAA-R residues have been mutated in knock-in mice, which subsequently lose sensitivity to general anesthetics, thus verifying the important role of GABAA-Rs in anesthetic action.12
Alcohol action and GABAA-Rs
Ethanol does enhance GABAA-Rs and glycine receptors, much like longer chain alcohols (e.g., n-octanol), but at very high (anesthetic) concentrations. We need to distinguish between these anesthetic effects of high-dose EtOH and longer chain alcohols and the intoxicating effects of lower doses, such as those encountered in the human brain during social drinking, because evidence suggests that effects of low- and high-dose EtOH involve (at least) two distinct targets in the brain. We suggest that these receptors that are sensitive to EtOH levels resulting from one glass of wine (3-30 mM) are certain subtypes of GABAA-R, the δ subunit-containing extrasynaptic GABAA-R that mediate tonic inhibition.15,16 One major target of anesthetic doses of EtOH (30-100 mM) is in the transmembrane domain of GABAA-Rs and glycine receptors.13,17,18
Evidence suggests that EtOH has a second site of interaction with GABAA-R that is relevant to its pharmacological actions at low doses as produced in humans by one glass of wine.16 Studies on a naturally-occurring allele in the rat GABAA-R α6 subunit suggests that the residue, α4/6R100, is involved in EtOH action as well as its blockade by the benzodiazepine ligand, Ro15-4513.17,19,20 These results are consistent with our observations that the α4/6β3δ subtypes of GABAA-R are more sensitive to EtOH (low mM) than other subtypes, including the γ2 subunit or the β1/2 subunits.15 Moreover, this is consistent with the observation by many that EtOH at low mM concentrations enhances GABAA-R-mediated tonic inhibitory currents in cells expressing the δ GABAA-R subunit.19,21-23 This hypothesis remains controversial,24 as the expression of recombinant δ subunits is very difficult and thus hardly studied, and the brain slice/neuron work is still in its infancy; however, we believe the hypothesis will be accepted as it becomes better understood. Our preliminary investigations of domains within the β3 and δ subunits that interact in α4/6β3δ subtypes implicate residues in the β3 subunit that are homologous to the benzodiazepine binding site involving αR100, which produces an EtOH-sensitive binding site for certain benzodiazepines, like Ro15-4513, that would be located at the α+/β- interface in the extracellular domain. Future work will determine whether this model has any basis in reality.
Methods for identification of amino acids involved in specific functions of proteins, including allosteric modulators
Chimeras and site-directed mutagenesis
An often utilized and frequently successful technique is to compare polypeptide sequences of homologous subunits showing varying sensitivity to drugs. For example, Mihic et al.13 compared glycine receptors (sensitive to isoflurane and high-dose EtOH) with GABAA-R ρ subunits (insensitive to these modulators). One makes chimeric cDNAs that differ in the domain of interest and determine which residues are critical. This is followed by site-directed point mutations in suspected residues until the critical one(s) is identified. On the negative side, identification of residues that determine modulatory drug sensitivity by mutagenesis alone may be allosterically coupled to the function rather than involved in binding per se. Thus, the initial observation requires confirmation by secondary approaches. This could involve studying a number of mutations for the implicated residue to discover the shape and properties the “pocket” might show, or attempting a covalent couple of an analogue of the anesthetic containing a sulfhydryl reagent to the cysteine-substituted candidate residue.25-27
Affinity labelling
The use of affinity labels is more likely to identify residues involved in a binding pocket, because the ligand has to have reasonable affinity for the target protein and a chemical group that will bind the protein covalently. The ligand will label residues in close contact when it is bound to the receptor. To maximize specificity, the chemically reactive group is often stable until chemically activated, e.g., by ultraviolet light.28 Nevertheless, some uncertainty arises from possible movement of the activated ligand from the binding site to nearby areas, depending on its lifetime and relative reactivity. Thus, additional evidence consistent with identification of the labelled residues as binding site constituents is also useful here. Making the affinity label radioactive can allow identification of the subunit polypeptide-carrying binding sites on SDS gels and can aid in sequencing the peptide. This binding must be prevented by excess non-radioactive ligand, and its relevance to the drug receptor can be supported by evidence of allosteric modulation of its binding by other receptor ligands. Another advantage of affinity labelling is the fact that the results are unbiased by having to guess in advance which residues might be important, as required with mutagenesis. We succeeded in using this technique for anesthetic binding to GABAA-R.29
Major findings
Site-directed mutagenesis results
Wingrove et al. (1994)30 found that modulation of GABAR by the anticonvulsant/anxiolytic loreclezole depended on the nature of the β subunit (β2/β3 ≫ β1). Belelli et al. (1997)31 extended this β subunit selectivity to the intravenous anesthetic, etomidate, a chemical analogue of loreclezole, and demonstrated that the β selectivity was due to residue 265, which is N in both β2 and β3 (sensitive to etomidate) and differed from β1 (S, insensitive to etomidate) as well as the insect β (Rdl, which has M, insensitive to etomidate). They31 showed that a single amino acid in GABAA-R, β2N265, (βM2-15’) was responsible for the selectivity of β2 and β3 over β1 for the modulatory drugs, loreclezole and etomidate. This β selectivity was found to extend to the related intravenous anesthetic, propofol.32
Mihic et al. (1997)13 used the anesthetic-insensitive GABAA-R ρ subunit sequence and the anesthetic-sensitive glycine receptor α and GABAR α and β subunits to identify, by construction of chimeras and point mutations, some amino acid residues critical for modulation of GlyR and GABAA-R by volatile anesthetics and long-chain alcohols, implicating the M2-15’ and M3-4’ residues in both the α and β subunits of GABAA-R. Note that the M2-15’ residue is the same as the one found simultaneously by Belelli et al. (1997).31
Carlson et al. (2000)14 took a similar approach to identify a single amino acid at the entrance to M1(-2’) in GABAA-R α and β, i.e., β2G229, which when mutated to larger (e.g., F as in ρ) or charged residues gave significantly reduced sensitivity to modulation of binding and function by steroids, etomidate, propofol, and barbiturates, with enhanced direct gating by these drugs. Thus, it appeared that larger than normal amino acids at this position took the place of anesthetics in partially enhancing GABA, so that the extent of enhancement by the exogenous anesthetic ligand was reduced.
In addition to the residues identified by Mihic et al. (1997)13 for anesthetic doses of EtOH enhancement of glycine receptors (transmembrane α1S267[M2-15’]), Daryl Davies and Ron Alkana found that residues in the extracellular domain loop 2 (α1A52) are also needed33 and proposed to be in the same pocket as M2-15’. These workers also went on to show that modification of loop 2 in glycine receptor α1 subunits or in GABAA-R γ2 subunit, such as replacement by the loop 2 10-residue peptide fragment from the GABAA-R δ subunit, increases EtOH sensitivity about tenfold, as measured in oocyte recordings.34
Hosie et al. (2006)35 constructed chimeras replacing steroid-sensitive α1 and β2 subunits of the GABAA-R M1 – M2 residues with those from the steroid-insensitive insect Rdl clone; this reduced steroid modulation, potentiation of GABA, and direct activation of αβγ receptors when α1 was mutated, but not β2. Steroid modulation was shown to depend on the nature of several residues in the transmembrane helix. They provided evidence that α1Q242(M1-10’) and α1N408 and Y411 in M4 (bovine numbering)36 could provide anchor points to bind the two ends of the steroid molecule. Also, α1T237(M1-5’) (bovine numbering) and βY284 were shown to be essential for direct activation, and protein structural modelling suggested that they were located near each other in a single intersubunit pocket at the βM3-αM1 interface.35 This possible model is discussed further below.
Mouse knock-ins
The results of Belelli et al. (1997)31 led to the production of knock-in mice with point mutations in this single GABAA-R subunit βM2-15’ residue. Both the β2N265S (replaced with the β1 residue),37 which showed less sensitivity to the sedative action of etomidate, and the β3N265 M (replaced with the anesthetic-insensitive insect residue),38 which showed less sensitivity to the immobilization action of etomidate and propofol, provided strong evidence for the role of GABAA-Rs in anesthetic action. Furthermore, a subunit composition-dependent pharmacology was established, presumably due to an anatomical correlate of anesthetic sensitivity. In other words, the β3-containing GABAA-Rs and the circuits in which they function are involved in the anesthetic actions of etomidate, while the β2-containing GABAA-Rs are not but are involved in some of the sedative and other actions of etomidate; β1-containing GABAA-Rs are not involved in anesthetic actions of etomidate.12
Harrison et al. went on to test the α1S270H mutation in knock-in mice for reduced sensitivity to anesthetics. Hall et al. (2004)39 found a reduced sensitivity to isoflurane in enhancing GABAA-R current, with increased probability of opening for the mutant channels in absence of anesthetic. Elsen et al. (2006)40 found altered responses to volatile anesthetics in vivo, including reduced time of anesthesia and abnormal seizure-like behaviours on coming out of anesthesia. This is not strong evidence for an anesthetic site of action. However, the α1S270H (M2-15’) knock-in mice had a sickly phenotype, interpreted as likely due to the left-shift in the GABA dose-response curve and a corresponding excessive GABAergic inhibition in vivo.39 A compensatory second knock-in point mutation, α1L277A, introduced into the S270H mouse restored normal neuronal and in vivo excitability levels, normal GABA sensitivity, and normal health41 and allowed demonstration that these animals exhibited less than normal responses in some assays of volatile anesthetic42 and high (anesthetic)- dose EtOH effects.43
Affinity labelling with azietomidate
We have used the technique of photoaffinity labelling, synthesizing a photo-incorporable analogue of etomidate with anesthetic efficacy, R(+)azietomidate, and then radiolabelling it.44 This ligand [3H]azietomidate was successfully employed to photolabel benzodiazepine affinity-purified GABAA-R proteins from cow brain and identify two amino acids as sites of attachment.29 The team led by Keith Miller at Massachusetts General Hospital synthesized a photo-incorporable anesthetic, R(+)azietomidate, a close analogue of etomidate, and demonstrated both bio-activity as well as photoaffinity labelling capability.44 Using detergent-solubilized membranes that retained the allosteric modulation of GABAA-R radioligand binding by etomidate, the Richard Olsen lab purified to homogeneity the GABAA-R protein from bovine cerebral cortex and photoaffinity labelled the pooled protein (several hundred pmol) with [3H]azietomidate, leading to a single peak of radioactive protein at ca. 55 kDa, which was shown by mass spectrometry and Western blotting to contain GABAA-R α1, α2, α3, α5, β1, β2, β3, and γ2 (and a trace of α4) subunits. Edman degradation microsequencing of proteolytic fragments purified on high performance liquid chromotogrpahy (HPLC) by the Jon Cohen lab allowed identification of the labelled amino acids. The residues that were labelled do not appear to be nonspecifically tagged. The same results have now been obtained on numerous occasions; alkyl azides are not particularly reactive with Met residues, and several more reactive residues as well as two other methionines in the region near our two identified amino acids were not reactive.
The residues labelled were M236 in the M1 domain of the alpha subunits (α1, 2, 3, and 5 were all the same in the relevant peptides) and M286 in the M3 domain of the beta subunits (also identical in β1, 2, and 3). The attachment sites for etomidate were thus identified as αM1-11’ (M236) and βM3-4’ (M286) (Table 1).29 The latter is the same as one of the two identified by Mihic et al. for anesthetic alcohols and volatile agents.13 The former had not been described previously for anesthetic interactions. Figure 1 shows a 3-D model of the heteropentameric GABAA-R with the GABA and BZ sites at subunit interfaces in the extracellular domain and etomidate sites in the transmembrane domain.29 The BZ sites are modified GABA sites located at a different subunit interface, that is, homologous residues corresponding to agonist binding pocket loops in the protein at the two β/α interfaces for GABA binding are involved in BZ binding at the single α/γ interface.8,11,45

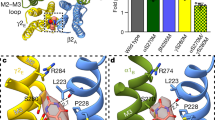
Three-dimensional views of the γ-aminobutyric acid type A receptors (GABAAR) homology model from different perspectives (A,B) and helical wheel representation of the transmembrane regions at the etomidate binding domain at the β/α interface (C). A) Three-dimensional view of the GABAAR homology model (β3, cyan; α1, yellow; γ2, green) from a perspective outside the membrane looking down the channel or B) from a perspective parallel to the membrane surface. Modified from Li et al. (2006)29 in which our homology model is based roughly on the nicotinic acetylcholine receptor cryo-EM derived structure (PDB:2BG9),47 modified by helical alignment data from Jansen & Akabas (2006).51 Pockets in the vicinity of the etomidate (maroon), GABA (purple), and benzodiazepine (red) binding sites are shown as Connolly surfaces. C) Helical wheel representation of the GABAAR β/α interface in the trans-membrane domain illustrating the proposed binding site for etomidate (modified from Li et al., 2009).36 The model illustrates the orientation of residues from a homology model built on the nicotinic acetylcholine receptor cryo-EM derived structure,47 with the residues in αM1 and βM3 photolabelled by [3H]azietomidate (circled residues in green) contributing to a common binding pocket at the β/α interface. Also included: the position in βM2 (N265) that functions as a determinant of etomidate/azietomidate anesthetic potency in vivo (pink)37,38,62; the residues in αM1 and βM3 identified as sensitivity determinants for direct activation by neurosteroids (boxed residues in yellow)35; and the positions in αM1 and βM3 that can form intersubunit cross-links when mutated to Cys (red and orange)52,58
Interaction of other general anesthetics with azietomidate labelling sites
The Table summarizes the studies with other chemical classes of general anesthetics. The intravenous agent, propofol, with a very similar pharmacological profile was able to inhibit the labelling of GABAA-R with azietomidate at anesthetic concentrations, but only partially, even when present in large excess over the etomidate. This partial inhibition was evident on both labelled residues, as shown by sequencing. The binding is not mutually exclusive, although it could involve overlapping sites. We conclude that it indicates an allosteric interaction.46 A similar partial inhibition was seen for a series of pharmacologically active barbiturates, also apparently allosteric.46 In addition, the anesthetic neurosteroids at appropriate concentrations did not inhibit azietomidate labelling but rather enhanced it. This proves that the steroid site does not coincide with the etomidate binding pocket and interacts allosterically.36 On the other hand, the volatile agent, isoflurane, showed a smooth inhibition curve down to zero and thus could possibly reflect competitive inhibition. However, the alcohols, n-octanol (10-1000 μM) and EtOH (1-1000 mM), failed to interact with azietomidate labelling, neither enhancing nor inhibiting.46 We have suggested another site for pharmacological actions of EtOH at low concentrations on GABAA-R.16
Homology modelling
Computer-enhanced electron microscope images of the Torpedo nicotinic acetylcholine receptor47 and the water-soluble acetylcholine binding protein48 allowed homology modelling of the GABAA-R and positioned the two etomidate labelled residues sufficiently close to each other (about 10 Ǻ) in the transmembrane domain to allow us to propose a single binding pocket at the interface between αM1 and βM3 (Li et al., 2006,29 Electronic Supplementary Material). This intersubunit etomidate binding pocket (Fig. 2) would be present in two (identical) copies per pentamer at the same subunit interfaces (β+/α-) where GABA binding occurs in the extracellular domain. This seems a suitable position for allosteric modulation, especially relevant to the quaternary twist allosteric model for the pentameric ligand-gated ion channel receptors.49 The presence of only one kind of etomidate site29 suggests that both enhancement of GABA and direct gating by etomidate involve this one site. This is consistent with modelling of functional data following the Monod-Wyman-Changeux 2-state allosteric model.50

The intersubunit anesthetic binding pocket in the γ-aminobutyric acid type A (GABAA) receptor transmembrane domain identified by photoaffinity labelling with [3H]azietomidate.29 The identified residue, M236, in the M1 domain of the α subunit is situated nearly adjacent to the identified residue, M286, in the M3 domain of the β subunit. Reproduced with permission from: Li GD, Chiara DC, Sawyer GW, Hussain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabelling with an etomidate analog. J Neurosci 2006; 45: 11599-605
Jansen & Akabas51 used “cysteine scanning accessibility measurements” and cross-linking to align the M2 and M3 helices of the nicotinic acetylcholine receptor and used homology GABAAR to allow a fixed setting of the helical wheels relevant to our proposed etomidate site.29,36 Bali et al.52 continued in this vein using cysteine substituents to align the M1 and M3 helices of GABAAR, refining the helical wheel orientations (Fig. 1C). This showed our two etomidate residues, αM1-11’ and βM3-4’, to be very close and consistent with a single intersubunit anesthetic binding pocket at the α/β interface. One can also see that the βM2-15’ is not directly adjacent to the etomidate binding pocket but also is not very distant, and so it could possibly move into closer contact with αM1-11’ and βM3-4’. On the other hand, the two residues implicated in steroid direct gating action35 (Fig. 1C) are on different sides of the helices and could not be part of a single binding pocket due to the distance between them. Those residues implicated in steroid enhancement of GABA are not shown on this helix but would not overlap with the etomidate enhancement site we defined.
Biochemistry and electrophysiological testing of models derived from residue identification and modelling
Work in the lab of Harrison25 as a follow-up to Mihic et al.13 showed that the GABAA-R αM2-15’ involvement for volatiles was sensitive to the volume of the amino acid at that position, consistent with a binding pocket. Follow-up work in the lab of Harris26 showed that an alcohol sulfhydryl analogue, propanethiol, could be covalently attached to αM2-15’ when substituted by cysteine, leading to persistent enhancement of GABA responses. This was even more dramatically demonstrated for β2M2-15’, which could be modified by the alcohol, octane methanethiosulfonate, when mutated to cysteine to produce a covalent attachment showing irreversible potentiation53 consistent with a long-chain alcohol binding site. The binding of the octanol analogue could be inhibited by n-octanol, and the modification was able to alter modulation of GABA function by butanol and isoflurane, with some reduction of modulation by alphaxalone and flunitrazepam, but not pentobarbital.53 Homology modelling in which GABAA-R transmembrane helices were aligned, as found in the nicotinic acetylcholine receptor,47 suggested that these two residues, α or β M2-15’ and M3-4’, could be found in a single intrasubunit pocket.13,18 This supports the idea that the M2-15’ residue could be a binding site for long-chain alcohols and maybe volatile agents and could be sensitive to allosteric modulation by etomidate, propofol, and possibly other anesthetics. However, residues identified by mutagenesis alone could also be involved “merely” in allosteric transduction.
Work in the lab of Olsen as follow-up to Carlson et al. (2000)14 found mutations at this M1 residue, α or βM1(-2’), to alter rates of desensitization to GABA agonists and allosteric modulators.54 Mutations of β2M1(-2’) to a series of residues differing in volume showed altered gating kinetics consistent with a binding pocket.55 These mutagenesis studies also showed distinctive effects on anesthetic direct gating vs enhancement of GABA, consistent with two separate sites of action. However, the lack of chemical specificity for the modulators involved is more consistent with an allosteric transduction site than a binding pocket.
Mutagenesis analysis of the two etomidate binding residues of Li et al. (2006)29 has been studied by our lab56 and collaborator, Stu Forman. The first study replaced αM236 or βM286 with trp. Either mutation led to reduced allosteric modulation of GABA by etomidate, with increased spontaneous openings, GABA left-shift, and increased direct gating by etomidate,57 which we interpret as being consistent but not conclusive with the theory that the residues are in a binding pocket. The second study replaced the two etomidate residues with cysteine. With radioligand binding, we found that large but not small sulfhydryl reagents reacted with the α1M236 (M1-11’) in a manner that irreversibly prevented etomidate modulation of GABAA-R ligand binding; furthermore, the reaction with the sulfhydryl reagents was protected by excess etomidate in the tube.56 Stewart & Forman (2009)58 used electrophysiology with α1β2γ2 GABAA-R to show that mutation β2M286C (M3-4’) and β2N265C (M2-15’) could react with the sulfhydryl reagent, p-Cl mercuribenzene sulfonate, to eliminate etomidate modulation of channel function, but that excess etomidate protected the M286 residue, but not the N265 residue, from reaction with the sulfhydryl reagent. This is consistent with the etomidate residues identified by affinity labelling as being part of an anesthetic binding pocket.
Summary of residues in GABAA-Rs identified as necessary for anesthetic action
Homology modelling with the data of Unwin47 as a template suggested that the two etomidate binding residues could be part of a single class (two copies per pentamer) of intersubunit binding pocket (Fig. 2).29 This is also consistent with a single site for etomidate direct gating and modulation of GABA current, as suggested by Rüsch et al. (2004).50 Crosslinking studies of cysteine-substituted helical residues in the Akabas lab51,52 were totally consistent with this model, because residues that can crosslink between αM1 and βM3 position our etomidate residues, αM236 and βM286, nearly adjacent to each other (Fig. 1C).29,36 The transmembrane intersubunit pocket (Fig. 2) appears well situated to allosterically modulate the quaternary twist conformational change produced to gate the channel after binding of agonist to its site in the same β/α subunit interface of the extracellular domain 50 Ǻ directly above (Fig. 1). Nevertheless, the general anesthetics, desflurane, halothane, and propofol, were shown to inhibit channel function59 of another pentameric ligand-gated ion channel, the bacterial GLIC channel,60 and binding of these anesthetics was shown in x-ray crystal structures to occur at intrasubunit pockets in the transmembrane domain (P-J Corringer, MAC2010 symposium).
The intersubunit pocket between βM3 and αM1 for etomidate seems highly likely at this point. It remains unclear whether the other residue labelled by Mihic et al. (1997)13 at M2-15’ is part of this pocket. This residue was not affinity labelled by azietomidate. In our structural model, it is located sufficiently close to potentially participate in the binding pocket (Fig. 1C).29,36 Nevertheless, cysteine substitution of the three residues and inactivation by cysteine reagents revealed that etomidate occupancy was able to protect the cysteine reaction at α1M236C and β2M286C, but not at β2N265C, suggesting that the first two residues, but not the last, are in the etomidate binding pocket.58 This had been suggested earlier by Bali & Akabas61 who showed that propofol occupancy could protect from cysteine reagent inactivation at αM286C, but not at αN265C. The M2-15’ residue is clearly very important for anesthetic action. If it is part of the intersubunit pocket (αM1/βM2/βM3), it is unlikely that it would also be part of an intrasubunit pocket (αM1/αM2/αM3 or βM1/βM2/βM3), but this remains to be seen. It could differ for the different chemical classes of general anesthetics. There is evidence26 that propanol can bind to the αM2-15’ and octanol53 can bind to the βM2-15’, but these compounds do not interact with the etomidate binding residues, αM1-11’ and βM3-4’ (Li et al.).
Hosie et al.35 identified residues in αM1 and αM4 needed for neurosteroid enhancement of GABAA-R and two residues in αM1 and βM3 needed for direct steroid gating, which were proposed to be part of a single intersubunit anesthetic steroid site. However, those two residues are not positioned near each other in the helical wheels generated by models of the intersubunit etomidate site consistent with the cysteine substitution cross-linking data29,52 and consistent with our demonstration that the GABAA-R steroid ligands do not inhibit azietomidate labelling but rather enhance the binding in a pharmacologically specific manner.36 Our study of additional chemical classes of general anesthetics (Table 1) showed that propofol inhibited azietomidate labelling, but only partially, and thus probably allosterically.46 Likewise, barbiturates partially inhibited azietomidate labelling in a pharmacologically and stereo-specific manner, thus were probably allosteric. The volatile agent, isoflurane, gave a complete and possibly competitive inhibition of azietomidate labelling, while two alcohols, n-octanol and EtOH, at pharmacologically active concentrations gave no inhibition or enhancement of azietomidate labelling of brain GABAA-R.46 Taken together, the data on [3H]azietomidate labelling suggest that we have directly identified amino acids in the contact point and binding pocket for a general anesthetic in a relevant biological target of action, the GABAA-R protein in the mammalian brain. This information and future studies on the topic should provide a path to development of safer, more effective, and more selective general anesthetics.
References
Campagna JA, Miller KW, Forman SA. Mechanisms of actions of inhaled anesthetics. N Engl J Med 2003; 348: 2110-24.
Hemmings HC Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci 2005; 26: 503-10.
Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nature Rev Neurosci 2008; 9: 370-86.
Macdonald RL, Olsen RW. GABAA receptor channels. Ann Rev Neurosci 1994; 17: 569-602.
Luddens H, Korpi ER, Seeburg PH. GABAA/benzodiazepine receptor heterogeneity: neurophysiological implications. Neuropharmacology 1995; 34: 245-54.
Mody I, Pearce RA. Diversity of inhibitory neurotransmission through GABAA receptors. Trends Neurosci 2004; 27: 569-75.
Li G, Chang CS, Olsen RW. (2005) Anesthetic sites on GABAA receptors. In: Mashimo T, Ogli K, Uchida I (Eds). Basic and Systemic Mechanisms of Anesthesia. Amsterdam, Elsevier; Int Cong Ser 2005; 1283: 61-6.
Olsen RW, Sieghart W. International Union of Pharmacology LXX. Subtypes of γ-aminobutyric acidA receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 2008; 60: 243-60.
Jacob TC, Moss SJ, Jurd R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nature Rev Neurosci 2008; 9: 331-43.
Olsen RW, Fischer JB, Dunwiddie TV. Barbiturate enhancement of γ-aminobutyric acid receptor binding and function as a mechanism of anesthesia. In: Roth SH, Miller KW, editors. Molecular and Cellular Mechanisms of Anesthetics. New York: Plenum Medical Book; 1986. p. 165-77.
Olsen RW, Sapp DM, Bureau MH, Turner DM, Kokka N. Allosteric actions of central nervous system depressants including anesthetics on subtypes of the inhibitory γ-aminobutyric acidA receptor-chloride channel complex. Ann N Y Acad Sci 1991; 625: 145-54.
Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 2004; 5: 709-20.
Mihic SJ, Ye Q, Wick MJ, et al. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature 1997; 389: 385-9.
Carlson BX, Engblom AC, Kristiansen U, Schousboe A, Olsen RW. A single glycine residue at the entrance to the first membrane-spanning domain of the γ-aminobutyric acid type A receptor β2 subunit affects allosteric sensitivity to GABA and anesthetics. Mol Pharmacol 2000; 57: 474-84.
Wallner M, Hanchar HJ, Olsen RW. Ethanol enhances alpha 4/beta 3/delta and alpha 6/beta 3/delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci U S A 2003; 100: 15218-23.
Olsen RW, Hanchar HJ, Meera P, Wallner M. GABAA receptor subtypes: the “one glass of wine” receptors. Alcohol 2007; 41: 201-9.
Wallner M, Hanchar HJ, Olsen RW. Low-dose alcohol actions on α4β3δ GABAA receptors are reversed by the behavioral alcohol antagonist Ro15–4513. Proc Natl Acad Sci U S A 2006; 103: 8540-5.
Yamakura T, Bertaccini E, Trudell JR, Harris RA. Anesthetics and ion channels: molecular models and sites of action. Ann Rev Pharmacol Toxicol 2001; 41: 23-51.
Hanchar HJ, Dodson PD, Olsen RW, Otis TS, Wallner M. Alcohol-induced motor impairment caused by increased extrasynaptic GABAA receptor activity. Nat. Neurosci 2005; 8: 339-45.
Hanchar HJ, Chutsrinopkun P, Meera P, et al. Ethanol potently and competitively inhibits the binding of the alcohol antagonist Ro15–4513 to α4/6β3δ GABAA receptors. Proc Natl Acad Sci U S A 2006; 103: 8546-51.
Wei W, Faria LC, Mody I. Low ethanol concentrations selectively augment the tonic inhibition mediated by δ subunit-containing GABAA receptors in hippocampal neurons. J Neurosci 2004; 24: 8379-82.
Liang J, Zhang N, Cagetti E, Houser CR, Olsen RW, Spigelman I. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. J Neurosci 2006; 26: 1749-58.
Jia F, Chandra D, Homanics GE, Harrison NL. Ethanol modulates synaptic and extrasynaptic GABAA receptors in the thalamus. J Pharmacol Exp Ther 2008; 326: 475-82.
Lovinger DM, Homanics GE. Tonic for what ails us? High-affinity GABAA receptors and alcohol. Alcohol 2007; 41: 139-43.
Jenkins A, Greenblatt EP, Faulkner HJ, et al. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci 2001; 21: RC136.
Mascia MP, Trudell JR, Harris RA. Specific binding sites for alcohols and anesthetics on ligand-gated ion channels. Proc Natl Acad Sci U S A 2000; 97: 9305-10.
Tan KR, Gonthier A, Baur R, Ernst M, Goeldner M, Sigel E. Proximity-accelerated chemical coupling reaction in the benzodiazepine-binding site of γ-aminobutyric acid type A receptors: superposition of different allosteric modulators. J Biol Chem 2007; 282: 26316-25.
Kotzyba-Hibert F, Kapfer I, Goeldner M. Recent trends in photoaffinity labeling. Angew Chem Int Ed Engl 1995; 34: 1296-312.
Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci 2006; 26: 11599-605.
Wingrove PB, Wafford KA, Bain C, Whiting PJ. The modulatory action of loreclezole at the gamma-aminobutyric acid type A receptor is determined by a single amino acid in the beta 2 and beta 3 subunit. Proc Natl Acad Sci U S A 1994; 91: 4569-73.
Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anesthetic etomidate with the γ-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci U S A 1997; 94: 11031-6.
Siegwart R, Jurd R, Rudolph U. Molecular determinants for the action of general anesthetics at recombinant α2β3γ2 γ-aminobutyric acidA receptors. J Neurochem 2002; 80: 140-8.
Crawford DK, Trudell JR, Bertaccini EJ, Li K, Davies DL, Alkana RL. Evidence that ethanol acts on a target in Loop 2 of the extracellular domain of alpha1 glycine receptors. J Neurochem 2007; 102: 2097-109.
Perkins DI, Trudell JR, Crawford DK, Asatryan L, Alkana RL, Davies DL. Loop 2 structure in glycine and GABA(A) receptors play a key role in determining ethanol sensitivity. J Biol Chem 2009; 284: 27304-14.
Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature 2006; 444: 486-9.
Li GD, Chiara DC, Cohen JB, Olsen RW. Neurosteroids allosterically modulate binding of the anesthetic etomidate to γ-aminobutyric acid type A receptors. J Biol Chem 2009; 284: 11771-5.
Reynolds DS, Rosahl TW, Cirone J, et al. Sedation and anesthesia mediated by distinct GABAA receptor isoforms. J Neurosci 2003; 23: 8608-17.
Jurd R, Arras M, Lambert S, et al. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J 2003; 17: 250-2.
Hall AC, Rowan KC, Stevens RJN, Kelley JC, Harrison NL. The effects of isoflurane on desensitized wild-type and α1(S270H) γ-aminobutyric acid type A receptors. Anesth Analg 2004; 98: 1297-304.
Elsen FP, Liljelund P, Werner DF, Olsen RW, Homanics GE, Harrison NL. GABAA-R α1 subunit knockin mutation leads to abnormal EEG and anesthetic-induced seizure-like activity in mice. Brain Res 2006; 1078: 60-70.
Borghese CM, Werner DF, Topf N, et al. An isoflurane- and alcohol-insensitive mutant GABAA receptor alpha1 subunit with near normal affinity for GABA: characterization in heterologous systems and production of knock in mice. J Pharmacol Exp Ther 2006; 319: 208-18.
Werner DF, Swihart A, Rau V, et al. Inhaled anesthetic responses of recombinant receptors and knockin mice harboring α2(S270H/L277A) GABAA receptor subunits that are resistant to isoflurane. J Pharmacol Exp Ther 2010; DOI: 10.1124/jpet.110.170431.
Werner DF, Blednov YA, Ariwodola OJ, et al. Knockin mice with ethanol-insensitive alpha1-containing γ-aminobutyric acid type A receptors display selective alterations in behavioral responses to ethanol. J Pharmacol Exp Ther 2006; 319: 219-27.
Husain SS, Ziebell MR, Rusch D, et al. 2-(3-Methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate: a derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels. J Med Chem 2003; 46: 1257-65.
Sigel E, Buhr A. The benzodiazepine binding site of γ-aminobutyric acid type A (GABAA) receptors. Trends Pharmacol Sci 1997; 18: 425-9.
Li GD, Chiara DC, Cohen JB, Olsen RW. Numerous general anesthetics inhibit etomidate binding to GABAA receptors. J Biol Chem 2010; 285: 8615-20.
Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 2005; 346: 967-89.
Brejc K, van Dijk WJ, Klaassen RV, et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001; 411: 269-76.
Taly A, Corringer PJ, Grutter T, et al. Implications of the quaternary twist allosteric model for the physiology and pathology of nicotinic acetylcholine receptors. Proc Natl Acad Sci U S A 2006; 103: 16965-70.
Rusch D, Zhong H, Forman SA. Gating allosterism at a single class of etomidate sites on α1β2γ2L GABAA receptors accounts for both direct activation and agonist modulation. J Biol Chem 2004; 279: 20982-92.
Jansen M, Akabas MH. State-dependent cross-linking of the M2 and M3 segments: functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J Neurosci 2006; 26: 4492-9.
Bali M, Jansen M, Akabas MH. GABA-induced intersubunit conformational movement in the GABAA receptor α1M1-β2M3 transmembrane subunit interface: experimental basis for homology modeling of an intravenous anesthetic binding site. J Neurosci 2009; 29: 3083-92.
McCracken ML, Borghese CM, Trudell JR, Harris RA. A transmembrane amino acid in the GABAA receptor β2 subunit critical for the actions of alcohols and anesthetics. J Pharmacol Exp Ther 2010; DOI: 10.1124/jpet.110.170472.
Engblom AC, Carlson BX, Olsen RW, Schousboe A, Christiansen U. Point mutation in the first transmembrane region of the β2 subunit of the γ-aminobutyric acid type A receptor alters desensitization kinetics of γ-aminobutyric acid and anesthetic-induced channel gating. J Biol Chem 2002; 277: 17438-47.
Chang C, Olcese R, Olsen RW. A single M1 residue in the β2 subunit alters channel gating of GABAA receptor in anesthetic modulation and direct activation. J Biol Chem 2003; 278: 42821-8.
Li G, Olsen RW. A TM1 residue, α1M236, is important for a GABAA receptor anesthetic binding site. SfN 2007; 141.1 (abstract).
Stewart D, Desai R, Cheng Q, Liu A, Forman SA. Tryptophan mutations at azi- etomidate photo-incorporation sites on α1 or β2 subunits enhance GABAA receptor gating and reduce etomidate modulation. Mol Pharmacol 2008; 74: 1687-95.
Stewart D, Forman SA. Accessibility and etomidate protection of cysteine substitutions at azietomidate labeled residues in GABAA receptors. SfN 2009; 530.16 (abstract).
Weng Y, Yang L, Corringer PJ, Sonner JM. Anesthetic sensitivity of the Gloeobacter violaceus proton-gated ion channel. Anesth Analg 2010; 110: 59-63.
Bocquet N, Nury H, Baaden M, et al. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 2009; 457: 111-4.
Bali M, Akabas MH. Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol 2004; 65: 68-76.
Liao M, Sonner JM, Husain SS, et al. R(+) Etomidate and photoactivable R(+)azietomidate have comparable anesthetic activity in wild-type mice and comparably decreased activity in mice with a N265 M point mutation in the gamma aminobutyric acid receptor β3 subunit. Anesth Analg 2005; 101: 131-5.
Conflicts of interest
None declared.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is accompanied by an editorial. Please see Can J Anesth 2011; 58(2).
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Olsen, R.W., Li, GD. GABAA receptors as molecular targets of general anesthetics: identification of binding sites provides clues to allosteric modulation. Can J Anesth/J Can Anesth 58, 206–215 (2011). https://doi.org/10.1007/s12630-010-9429-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12630-010-9429-7