
Abstract
Apoptosis of endothelial cells related to homocysteine (Hcy) has been reported in several studies. In this study, we evaluated whether reactive oxygen species (ROS)-producing signaling pathways contribute to Hcy-induced apoptosis induction, with specific emphasis on NADPH oxidases. Human umbilical vein endothelial cells were incubated with 0.01–2.5 mM Hcy. We determined the effect of Hcy on caspase-3 activity, annexin V positivity, intracellular NOX1, NOX2, NOX4, and p47phox expression and localization, nuclear nitrotyrosine accumulation, and mitochondrial membrane potential (ΔΨ m). Hcy induced caspase-3 activity and apoptosis; this effect was concentration dependent and maximal after 6-h exposure to 2.5 mM Hcy. It was accompanied by a significant increase in ΔΨ m. Cysteine was inactive on these parameters excluding a reactive thiol group effect. Hcy induced an increase in cellular NOX2, p47phox, and NOX4, but not that of NOX1. 3D digital imaging microscopy followed by image deconvolution analysis showed nuclear accumulation of NOX2 and p47phox in endothelial cells exposed to Hcy, but not in control cells, which coincided with accumulation of nuclear nitrotyrosine residues. Furthermore, Hcy enhanced peri-nuclear localization of NOX4 coinciding with accumulation of peri-nuclear nitrotyrosine residues, a reflection of local ROS production. p47phox was also increased in the peri-nuclear region. The Hcy-induced increase in caspase-3 activity was prevented by DPI and apocynin, suggesting involvement of NOX activity. The data presented in this article reveal accumulation of nuclear NOX2 and peri-nuclear NOX4 accumulation as potential source of ROS production in Hcy-induced apoptosis in endothelial cells.
Similar content being viewed by others
Introduction
In 1962, Carson and Neill and Gerritsen et al. [1, 2] described severely increased levels of homocysteine (Hcy) in the urine of mentally retarded children. A few years later, this homocystinuria was linked to a severe genetic defect in the cysthationine β-synthase (CBS) gene [3]. Patients with this genetic defect were found to have occlusive cardiovascular disease even at a very young age [4]. Moderately increased plasma levels of Hcy, also termed hyperhomocysteinemia (HHC), are caused by several genetically determined enzyme deficiencies and the life style factor diet [5, 6]. Numerous in vivo and in vitro studies have also linked HHC to cardiovascular disease and suggest that it could be an independent risk factor for the development of atherothrombosis [7–10]. Whether Hcy itself is the culprit in the process of atherosclerosis or another Hcy-related substance, such as S-adenosylhomocysteine (SAH), remains to be elucidated. Since SAH is a potent inhibitor of methylation, it has been suggested that SAH rather than Hcy causes cellular damage [11]. However, the exact mechanisms and pathways for the induction of this damage remain to be elucidated. Furthermore, recent large-scale intervention trials using high-dose-B-vitamins to reduce plasma Hcy levels in cardiovascular patients have not shown an overall clinical benefit as yet [12, 13]. This raises further questions about the mechanisms underlying the association between HHC and cardiovascular disease, and also about the possible adverse effects of high-dose-B-vitamins as a means of reducing Hcy levels [14]. It therefore calls for more extensive research on the topic.
Growing evidence suggests that endothelial dysfunction plays a major role in vascular injury seen in HHC [15–17]. This has been observed in numerous animal models suggesting an involvement of increased oxidative stress with a depletion of bioactive nitric oxide and an impaired response to endothelium-derived hyperpolarizing factor [9, 18–21]. In vitro studies have also demonstrated that Hcy induces oxidative stress [22] and cell death [23–27] in endothelial cells. We have recently shown in cardiomyocytes that ischemia and Hcy-induced translocation of NADPH oxidase 2 (NOX2) to the nucleus, and subsequent production of nuclear reactive oxygen species (ROS) and apoptosis [28, 29]. In this study, we examined the role of specific NOX proteins in Hcy-induced endothelial cell death.
Materials and Methods
Cell Culture
HUVECs were isolated from umbilical cords and cultured in Medium 199 (BioWhittaker, Verviers, Belgium) supplemented with 10% heat-inactivated fetal calf serum (FCS; BioWhittaker), 10% heat-inactivated human serum (Sanquin, Amsterdam, The Netherlands), 5 μg/ml heparin (Leo Pharma BV, Breda, The Netherlands), 50 mg/ml endothelial cell growth factor (Sigma, St. Louis, MO, USA), 100 IU/ml penicillin (Yamanouchi Europe BV, Leiderdorp, Netherlands), and 100 μg/ml streptomycin (Radiopharma-Fisiopharma, Palomonte, Italy) at 37°C in a humidified 5% CO2/95% air atmosphere. Experiments were performed after two passages of the cells and at full confluence of the cells.
Antibodies and Chemicals
Monoclonal antibody 48 against NOX2 (gp91phox; dilution 1:28) was used [29–32], with Cy3-labeled goat-anti-rabbit-Ig (1:75) (Alexa Fluor; Leiden, Netherlands) as a secondary antibody. Although there is some sequence similarities between the binding site of the MAb48 and NOX5, no cross reactivity of the NOX2 antibody for NOX5 was found (courtesy of Dr KH Krause). Polyclonal α-Nitrotyrosine IgG (1:50) (Molecular Probes Inc, Eugene, OR, USA) was used as an antibody to measure nitrotyrosine residues which is an indicator for ROS production [33–35]. It was used with Cy5-labeled goat-anti-rabbit-Ig (1:50) (Jackson Immuno Research, West Grove, PA, USA) as a secondary antibody. The same procedure was followed for the immunofluorescent staining with goat-anti-p47phox antibody, goat-anti-Mox1 antibody (NOX1), and goat-anti-NOX4 (N-15) (all 1:50 dilution; all from Santa Cruz Biotechnology Inc, CA, USA), where we used Cy3-labeled donkey-anti-goat-Ig (1:40) (Alexa Fluor, Invitrogen) as a secondary antibody. Again, we co-stained for nitrotyrosine; however, now we used Cy5-labeled donkey-anti-rabbit (1:40) (DakoCytomation, Glostrup, Denmark) as a secondary antibody. Isotype and negative controls were employed to determine a-specific binding, which all were negative for staining (not shown). NOX-4 localization data were confirmed by employing another NOX-4 antibody (Novus Biologicals).
Cells were incubated with different concentrations (0.01–2.5 mM) of d,l-homocysteine (d,l-Hcy) (Sigma) in growth medium for 0–24 h at 37°C in a humidified 5% CO2/95% air atmosphere. 2.5 mM d,l-cysteine (Cys; Sigma) was also tested in the caspase-3 activity assay to distinguish between actual Hcy effect and the putative effect of a reactive thiol group [36].
Measuring Hcy Concentration in Growth Medium
Because several studies have suggested that only the l-form of Hcy is bioactive and the d-form has no cellular effect [37, 38], we determined the concentrations of d,l-Hcy and l-Hcy. d,l-Hcy was measured by HPLC and fluorescence detection [39]. Intra- and inter-assay coefficients of variation (CVs; std dev./average × 100%) were 1.8 and 3.5%, respectively. l-Hcy was measured by the Abbott IMx fluorescence polarization immunoassay (IMx; Abbott Laboratories, Abbott Park, IL, USA). Intra- and inter-assay CVs were less than 2 and 4%, respectively. The concentration of Hcy in culture medium was measured before incubation (t = 0) and after 6 h of incubation.
Determination of Intracellular S-Adenosylmethionine (SAM) and S-Adenosylhomocysteine (SAH)
Since it has been suggested that an increase in Hcy could cause cellular damage because of elevation of intracellular levels of SAH [11], we also determined the intracellular concentration of SAM and SAH in HUVECs after 6 h of incubation with d,l-Hcy. Liquid chromatography tandem mass spectrometry (LC–MS/MS) was performed as previously described [40]. The intra-assay CVs for SAM and SAH were 4.2 and 4.0%, respectively, and the inter-assay CVs were 7.6 and 5.9%, respectively.
Detection of Caspase-3 Activity
Cells were grown in 96-wells plates (20,000 cells/well). After treatment with Hcy and/or inhibitors (see below), cells were lysed and incubated with DEVD-rhodamine 110 substrate (Roche, Mannheim, Germany) for one hour at 37°C. Subsequently, the fluorescence of free rhodamine was determined using a microplate fluorescence reader (TECAN spectrafluor, Switzerland). The developed fluorochrome was proportional to the concentration of activated caspase-3 and was quantified by a calibration curve of diluted free rhodamine. Each condition was measured in triplicate/measurement (total of 3 measurements). Inhibitors used were the general caspase inhibitor Z-Asp-Glu-Val-Asp-fluoromethylketone (Z-VAD fmk, 25 μM final concentration, Alexis Biochemicals, Lausen, Switzerland), two NOX inhibitors (diphenylene iodonium, DPI, 10 μM final concentration, and apocynin, 100 μM final concentration, both from Sigma), and the mitochondrial ROS inhibitor rotenone, 50 μM final concentration (Sigma).
Flow Cytometry
Phosphatidylserine (PS) exposure was assessed with FITC-labeled human recombinant annexin V (Bender Med Systems, Vienna, Austria), while propidium iodide (PI; Bender Med Systems) was used to determine the permeability of the cellular membrane.
After treatment with Hcy, the cells were trypsinized and centrifuged at 400×g for 5 min at room temperature. Cells were then washed with serum-free M199, and re-suspended in serum-free M199 containing annexin V (1:40) for 30 min in the dark at 37°C in a humidified 5% CO2/95% air atmosphere. Shortly before measuring, propidium iodide (PI) was added to the cell suspension (1:40). Cells were measured using a FACSCalibur (Becton–Dickinson, San Jose, CA, USA). Results were analyzed by Cell Quest Pro software (Becton–Dickinson).
Western Blot Analysis
After treatment, HUVECs were harvested into modified ELB lysis buffer (250 mM NaCl, 0.1% Nonidet P-40, 50 mM HEPES pH 7.0, 5 mM EDTA, 0.5 mM DTT) with protease inhibitor cocktail (PIC, 1:40; Sigma). After determination of the protein concentration using the BCA protein assay kit (Pierce, Rockford, IL, USA), reducing sample loading buffer (0.25 M TRIS pH 6.8, sodium dodecyl sulfate (SDS), glycerol, 2-mercaptoethanol, bromophenol blue) was added, and the samples were mixed and heated at 95°C for 10 min. 50 μg protein of each sample was subjected to SDS-PAGE, transferred to nitrocellulose membranes, and analyzed for NOX2 expression with monoclonal antibody 48 (1:250 dilution), followed by horseradish-peroxidase-conjugated rabbit-anti-mouse-immunoglobulin (RαM-HRP; 1:1,000 dilution; DakoCytomation, Glostrup, Denmark). The blots were visualized by enhanced chemiluminescence (ECL; Amersham Biosciences AB; Uppsala; Sweden). Staining was quantified with a charge-coupled device (CCD) camera (Fuji Science Imaging Systems; Düsseldorf, Germany) in combination with AIDA Image Analyzer software (Isotopenmessgeräte; Staubenhardt, Germany).
3D Immunofluoresence Microscopy
To measure intracellular expression of NOX1, NOX2, NOX4, p47phox, and formation of nitrotyrosine, cells were incubated with or without Hcy for 6 h in 4-well chamber slides (Nalge Nunc International, Naperville, IL, USA) and treated as described before [29]. The cells were analyzed by a 3I MarianasTM digital imaging microscopy workstation (Zeiss Axiovert 200M inverted microscope; Carl Zeiss, Sliedrecht, Netherlands), equipped with a cooled CCD camera (Cooke Sensicam, 1280 × 1024 pixels; Cooke Co, Tonawanda, NY, USA) and nanostepper motor (Z-increments: 10 nm). Visualization of NOX1, NOX2, NOX4, p47phox, and nitrotyrosine was performed with a 40× oil lens. The microscope, camera, and data viewing as well as analysis process were controlled by SlideBookTM software (version 4.0.8.1; Intelligent Imaging Innovations, Denver, CO, USA) which allowed both real-time and 3D data acquisition (confocal optical stacks).
Live Cell Analysis of H2O2 Generation
Since the presence of nitrotyrosine residues is an indirect marker for ROS production, we also determined the generation of H2O2 which was measured intracellular with 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) (Molecular Probes, Leiden, The Netherlands). CM-H2DCFDA, being nonpolar, diffuses passively into cells, where its acetate groups are cleaved by intracellular esterases, and is trapped within the cell. In this status, it provides a substrate for oxidation by H2O2, resulting in the production of a highly fluorescent intracellular product emitting fluorescence with intensity proportional to the level of intracellular H2O2 [41, 42]. HUVECs were grown in Delta T dishes (0.17 mm, clear; Bioptechs Inc.; Butler, PA, USA); after incubation with or without Hcy the cells were loaded with CM-H2DCFDA (10 μM) in ADS buffer (in mM: 116 NaCl, 5.3 KCl, 1.2 MgSO4·7H2O, 1.13 NaH2PO4·H2O, 20 HEPES, and 1 CaCl2, pH 7.4); and incubated for 15 min at 37°C. Next, cells were incubated in ADS buffer for 25 min at 37°C, allowing the oxidized CM-H2DCFDA to accumulate in the cells. Fluorescence microscopy was performed by a 3I MarianasTM digital imaging microscopy workstation with a 10× air objective as described above.
Live Cell Analysis of ΔΨm
Life cell imaging was used to visualize real-time alterations in mitochondrial membrane potential (ΔΨ m). Cells were loaded for 30 min at 37°C with 0.5 μM of 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1; Cell Technology Inc, Minneapolis, USA). In non-apoptotic cells, JC-1 exists as a monomer and is visible on the green channel (FITC). In healthy cells, these monomers accumulate as aggregates in the mitochondria because of existing ΔΨ m. These aggregates are visible on the red channel (CY3) when viewed with a fluorescence microscope. Cells were washed with PBS before viewing and subsequently analyzed by means of 3I MarianasTM digital imaging microscopy workstation as described above.
Statistics
Data are expressed as mean ± SEM. Statistics were performed with the SPSS statistics program (Windows version 11.5). To evaluate whether observed differences were significant, One Way ANOVA with Post Hoc Bonferroni tests or student t tests were used where appropriate. A P value (two sided) of 0.05 or less was considered significant.
Results
Concentrations of d,l-homocysteine (d,l-Hcy), L-homocysteine (L-Hcy), S-adenosyl Methionine (SAM), and S-adenosyl Homocysteine (SAH)
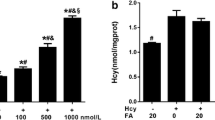
Since previous studies showed that only the l-form of Hcy is bioactive and that the l-form causes no cellular damage [37, 38], we measured the actual concentrations of both d,l-Hcy and l-Hcy before and after incubation of 2.5 mM d,l-Hcy. Before incubation on the cells, 43.3 ± 0.01% (n = 3) of the d,l-Hcy consisted of l-Hcy. The concentrations remained unaltered after a 6-h incubation period on HUVEC.
It has been suggested that not Hcy but an increase in SAH is the culprit in inflicting the damaging effects in HHC [11], since it acts as a potent methylation inhibitor. Therefore, we also measured the intracellular SAM/SAH levels resulting from incubation with 2.5 mM d,l-Hcy at 6 h. SAM increased significantly from 117 ± 2 nM (t = 0) to 184 ± 1 nM (t = 6-h incubation; P < 0.001). The increase in SAH was much higher, from 35 ± 2 nM (t = 0) to 521 ± 22 nM (t = 6-h incubation; P < 0.001; n = 3).
Effect of Hcy on Cell Death
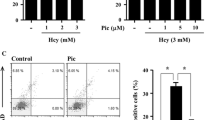
To test the effect of d,l-Hcy on caspase-3 activity as a reflection of apoptosis, concentrations ranging from 0.01 to 2.5 mM were tested, which are comparable to those in previous studies in cultured endothelial and smooth muscle cells [23, 24, 37]. Incubation for 3 h with 0.01, 0.1, and 1 mM d,l-Hcy induced a significant increase of caspase-3 activity by up to 15 ± 0.4% compared with controls. Incubation with 2.5 mM d,l-Hcy induced an increase of caspase-3 activity by 26 ± 2% (Fig. 1a; P < 0.001).

Effect of Hcy on caspase-3 activity a HUVECs were incubated for 3 h with the indicated concentrations of d,l-Hcy, whereafter caspase-3 activity was measured and compared with controls, where controls were set to 100% (n = 3; n is number of independent experiments with HUVEC of different donors). b Analysis of caspase-3 activity at different times of incubation (C control; Hcy 2.5 mM d,l-Hcy; n = 6). c Effect of Z-VAD-fmk on caspase-3 activity in HUVECs incubated with 2.5 mM d,l-Hcy for 6 h (n = 6). d Effect of 2.5 mM d,l-Hcy and 2.5 mM d,l-Cys for 6 h on caspase-3 activity in HUVECs (n = 6). e Flow cytometry analysis of HUVECs incubated with 2.5 mM d,l-Hcy for 6 h where single annexin V positivity as an indicator for PS exposure was determined (n = 6). f Flow cytometry analysis of HUVECs incubated with 2.5 mM d,l-Hcy for 6 h and annexin V/PI positivity as an indicator for plasma membrane permeability was determined (n = 6). All data represent mean and SEM
Over a 24-h evaluation period, the highest significant increase in caspase-3 activity induced by 2.5 mM d,l-Hcy (33 ± 8%, P = 0.001) was measured after 6 h of incubation (Fig. 1b). This increase was largely prevented by the simultaneous presence of the pan-caspase inhibitor Z-VAD FMK (Fig. 1c; P < 0.05). To determine whether it is actually Hcy that induced caspase-3 activity and not an unspecific effect of a reactive thiol group, we also incubated HUVECs with 2.5 mM d,l-cysteine. In contrast to Hcy, the presence of d,l-cysteine did not cause caspase-3 activation, indicating that the effect of Hcy is not due to an unspecific reactive thiol group effect (Fig. 1d).
To validate the caspase-3 results, we also determined putative phosphatidyl serine (PS) exposure on the outer leaflet of the plasma membrane by annexin V binding and membrane permeability for propidium iodide (PI) [36, 43]. Flow cytometric analysis of HUVECs that were incubated with 2.5 mM d,l-Hcy for 6 h also displayed a significant increase of single annexin V positive cells (8 ± 2%), part of which are early apoptotic (Fig. 1e; P < 0.01). Furthermore, we also found a significant increase by 11.3 ± 1% of annexin V/PI positive cells (Fig. 1f; P < 0.001), part of which are also (late) apoptotic.
All the following experiments were performed after 6-h incubation with 2.5 mM d,l-Hcy.
Alterations in ΔΨm
In order to analyze possible alterations in mitochondrial function, we monitored ΔΨm using the JC-1 probe. No Hcy-induced changes were detected in the morphology of the mitochondria, but an overall increased mitochondrial activity was observed in the Hcy-treated HUVECs (Fig. 2a, b). We quantified the sum intensity of the red, Cy3 signal, indicative for actively respiring mitochondria and indeed found a significant increase in ΔΨm (Fig. 2c; P = 0.003) in the Hcy-treated cells compared with control cells.

Effect of Hcy on mitochondrial membrane potential (ΔΨm) (a–c) analysis of mitochondrial membrane potential ΔΨm (magnification ×40) of HUVECs incubated with or without Hcy. Green (FITC) indicates JC-1 monomers normally present in the cytosol. With increasing ΔΨ, the monomers form aggregates and give a red (Cy3) fluorescence. a HUVECs incubated in growth medium alone. b HUVECs incubated with 2.5 mM d,l-Hcy for 6 h. Arrows indicate actively respiring mitochondria with increased ΔΨ. c Analysis of mean intensity of fluorescence Cy3 (red) signal, indicative for actively respiring mitochondria in HUVECs with or without 2.5 mM d,l-Hcy for 6 h where control was set to 100% (n = 3). Data represent mean and SEM (Color figure online)
Effect of Hcy on Intracellular NOX1, NOX2, NOX4 and P47phox Localization, Protein Nitrosylation, and H2O2 Generation
In a previous study on cardiomyocytes, we found that Hcy-induced nuclear NOX2 expression coinciding with nuclear nitrotyrosine residues, which ultimately resulted in apoptosis [29]. Therefore, we analyzed the effect of Hcy on NOX2 expression in endothelial cells. A constitutive presence of NOX2 in untreated HUVECs was revealed by Western Blot analysis, while incubation with 2.5 mM d,l-Hcy for 6 h resulted in a significant increase of NOX2 protein expression in whole cell lysates (Fig. 3; P < 0.001).

Effect of Hcy on NOX2 expression Western Blot analysis of whole cell lysates from HUVECs incubated with or without 2.5 mM d,l-Hcy for 6 h. The histogram represents a quantification of the bands from 3 separate experiments. 50 μg/sample was loaded. Equal loading was determined using actin staining. Control was set to 100%. Data represent mean and SEM
Next, the subcellular localization of NOX2 was analyzed by digital imaging fluorescence microscopy. In untreated HUVECs diffuse positivity of NOX2 was observed within the cell (Fig. 4a). However, the cells incubated with 2.5 mM d,l-Hcy additionally showed a significant increase in nuclear NOX2 localization (Figs. 4b, d, 5a; P < 0.001). To validate whether the observed NOX2 is present within the nucleus, we also performed 3D confocal stacking to acquire optical sections of the cells. Figure 4e, f shows an optical section after deconvolution of the out-of-focus light through the widest in-focus diameter of a nucleus. NOX2 is clearly present only in the nucleus after incubation with Hcy (Fig. 4f), but not without Hcy (Fig. 4e), and not in the perinuclear region (Fig. 4f). To assess whether NOX2 expression coincided with local ROS production, we also analyzed the presence of nitrotyrosine residues, which can be used as an indirect marker of ROS production. Only in HUVECs incubated with 2.5 mM d,l-Hcy for 6 h, nuclear nitrotyrosine formation was found, and it co-localized with NOX2 expression in the nucleus (Figs. 4c, d, 5b; P = 0.005). As depicted in Fig. 4c, f, in addition to nuclear nitrotyrosine, we found nitrotyrosine outside the nucleus (indicated with arrows in Fig. 4f). However, the latter did not co-localize with NOX2 expression.

Localization and quantification of NOX1, NOX2, NOX4, p47phox, and ROS in HUVECs incubated in the presence or the absence of Hcy Digital Imaging microscopy pictures (magnification ×40) of a HUVECs not incubated with Hcy. Red (Cy3) signal indicates NOX2 expression. Green (Cy5) indicates nitrotyrosine residues (arrows). b NOX2 (red, Cy3) expression in HUVECs incubated with 2.5 mM d,l-Hcy for 6 h. c Green (Cy5) nitrotyrosine formation, indicative for ROS production, in same HUVECs as figure b (arrows indicate cytoplasmic ROS). d Merged image of b and c, which shows yellow and orange coloration where NOX2 (red) and nitrotyrosine (Cy5) co-localize. e, f Optical section of a 3D stack after deconvolution from one nucleus incubated without (e) or with 2.5 mM d,l-Hcy (f) to determine exact location of NOX2 (red; Cy3) and nitrotyrosine (green; Cy5; arrows). g, h p47phox expression (red, Cy3) in HUVECs incubated without Hcy (g) or with Hcy (h) for 6 h. i, j Optical sections of a 3D stack after deconvolution from one nucleus incubated without (i) or with 2.5 mM d,l-Hcy (j) to determine exact location of p47phox (red; Cy3) and nitrotyrosine (green; Cy5); arrowheads indicate nuclear p47phox. k, l Optical section of a 3D stack after deconvolution from one nucleus incubated with 2.5 mM d,l-Hcy to determine exact location of NOX-1 (red; Cy3 in k) or NOX-4 (red; Cy3 in l) and nitrotyrosine (green; Cy5 in k, l); yellow co-staining is observed in l. In all pictures, blue DAPI staining for DNA (nuclei) is shown (Color figure online)

Quantification of nuclear NOX2, p47phox, nitrotyrosin residues, and cellular H2O2 in HUVECs incubated in the presence or the absence of Hcy Quantitative analysis of digital imaging microscopy of the effect of 6-h incubation with or without 2.5 mM d,l-Hcy on nuclear NOX2 expression (a), nuclear nitrotyrosine generation (b), nuclear p47phox expression (c), and cellular H2O2 levels (d). Sum intensity of fluorescence/nucleus was determined and depicted as % increase versus control where control was set to 100% (n = 4). Data represent mean and SEM
Subsequently, we determined putative p47phox localization in HUVECs incubated with 2.5 mM d,l-Hcy for 6 h. P47phox was diffusely present in untreated HUVECs (Fig. 4h). As seen in Fig. 4g–j, 5c, nuclear localization of p47phox increased significantly (P < 0.001) after incubation of HUVEC with Hcy. Analysis of optical sections acquired by 3D confocal stacking of HUVECs incubated with Hcy showed increased p47phox localization within the nucleus (arrowheads Fig. 4j), and particularly in the peri-nuclear region as compared to untreated cells (Fig. 4i). The p47phox partly co-localized with nitrotyrosine residue staining in both nucleus and peri-nuclear region (Fig. 4j). Minor diffuse p47phox staining was also observed in the remainder of the cell.
We also analyzed NOX1 and NOX4 expressions. Diffuse cellular localization of NOX1 was observed, which displayed only a non-significant minor increase after incubation with 2.5 mM d,l-Hcy in the peri-nuclear region (Fig. 4k; 7.3 ± 8.5%). NOX1 expression did not co-localize with nitrotyrosine residue staining. On the other hand, incubation of the cells with 2.5 mM d,l-Hcy for 6 h resulted in a significant increase of peri-nuclear NOX4 localization (38.9 ± 9.5%; P < 0.0001) compared with control cells, simultaneous with an increased peri-nuclear presence of nitrotyrosine (Fig. 4l). Neither NOX1 nor NOX4 was present within the nucleus.
The increase in Hcy-induced intracellular ROS, as suggested by the nitrotyrosine staining, was verified via assay of intracellular H2O2 in live cells, using CM-H2DCFDA as a substrate for H2O2-mediated oxidation. Indeed, after incubation with 2.5 mM d,l-Hcy for 6 h, a significant increase in intracellular H2O2 was determined (Fig. 5d; 67.7 ± 24.7%; P < 0.03).
NOX-derived ROS are Involved in Hcy-induced Apoptosis
To analyze whether the Hcy-enhanced NOX activities indeed had an influence on caspase-3 activity, we measured caspase-3 activity after incubation with 2.5 mM d,l-Hcy in the presence or the absence of the NOX inhibitors DPI and apocynin. In the presence of either DPI or apocynin, the Hcy-induced caspase-3 activity was significantly inhibited, suggestive for a causal contribution of NOX-mediated ROS production to apoptosis in Hcy HUVECs (Fig. 6a, b; P = 0.023, P = 0.013).

Effect of NADPH oxidase inhibitors on caspase-3 activity. Effect of DPI (a), apocynin (b), and rotenone (c) on caspase-3 activity after incubation with 2.5 mM d,l-Hcy for 6 h. Caspase-3 activity is depicted as percentile increase compared to control where control was set to 100% (n = 3). Data represent mean and SEM. The experiments given in panel c were performed with HUVEC derived from different donors than those described above in a, b
Mitochondria-derived ROS are Involved in Hcy-induced Apoptosis
Since it is known that mitochondrial ROS can be involved in the process of apoptosis, we also determined the effect of the mitochondrial ROS inhibitor rotenone on the induction of apoptosis. Indeed, after simultaneous incubation of HUVEC with 2.5 mM d,l-Hcy and rotenone, a significant decrease in caspase-3 activity (by 48.6%) was found as compared with incubation with Hcy alone (Fig. 6c; P < 0.01).
Discussion
Incubation of HUVECs with Hcy resulted in apoptosis of endothelial cells measured via activation of caspase-3 and surface expression of annexin V. In the present study, we demonstrated by digital imaging analysis an increased expression and translocation of NOX2 and p47phox to the nucleus, which coincided with nuclear nitrotyrosine accumulation. Furthermore, incubation with Hcy resulted in increased peri-nuclear NOX4 and p47phox localization, which also in part was accompanied by the accumulation of nitrotyrosine. Inhibition of NOX-mediated ROS production by DPI or apocynin inhibited caspase-3 activation, while inhibition of mitochondrial ROS production also inhibited apoptosis. It is unlikely that the Hcy effect on endothelial cells is due to a non-specific reactive thiol group effect, because cysteine did not induce caspase-3 activity or apoptosis.
Several studies have examined Hcy-induced apoptosis in endothelial cells of different origin and the possible involvement of ROS, whether or not derived from NADPH oxidase activity. Induction of apoptosis and increased caspase-3 activity has been observed in HUVEC [44–46] and rat heart microvascular EC [26]. Increased cellular ROS production by Hcy was found in HUVEC [24, 44, 46–48], in mouse [49] and rat aorta EC [50], and in mouse brain [51], rat renal microvascular EC [52], and rat heart microvascular EC [26, 53]. It was inhibited by DPI [44], apocynin [27, 51,] and by p22phox siRNA [50], suggestive of NADPH oxidase involvement. Recently, Bao et al. [44] reported that Hcy-induced apoptosis in endothelial cells indeed occurred via NADPH oxidase-related oxidative stress and subsequent p38MAPK signaling. However, to our knowledge, this is the first time that Hcy-induced nuclear NOX2 and peri-nuclear NOX4 accumulation coinciding with ROS production and apoptosis has been described in endothelial cells.
A study by Suhara et al. [24] revealed that 5 mM Hcy for 12 h induced endothelial cell apoptosis via NF-κB-mediated upregulation of Fas (CD95) expression and subsequent activation of the JNK pathway. Zhang et al. [25] found that incubation with 3 mM Hcy for 24 h induced apoptosis in endothelial cells through activation of the unfolded protein response (UPR) which in turn also caused activation of JNK [54]. Dong et al. [27], however, suggested that Hcy induced oxidative stress and apoptosis not only via a JNK-dependent mechanism but also via NADPH oxidase. They found in particular that 1 mM Hcy for 24 h induced apoptosis, intracellular ROS generation (determined with CM-H2DCFDA fluorescence intensity), as well as an increase in the p47phox subunit as shown by western blot analysis of whole cell lysates [27]. Furthermore, they determined that caspase-3 activity was inhibited by apocynin and/or JNK inhibitor II with no additional effect when added together, suggesting that they are part of the same Hcy-induced apoptosis pathway [27]. This was further corroborated by Bao et al. [44] who showed that atorvastatin inhibited NOX-induced ROS production in endothelial cells. Our data on nitrotyrosine accumulation concur with these findings, and add to them by showing the specific localization of NOX2 (nuclear) and NOX4 (peri-nuclear), which were accompanied by the accumulation of nitrotyrosine residues. As the Hcy-induced activation of caspase-3 is inhibited largely by the NOX inhibitors DPI and apocynin, it is likely that the nuclear and peri-nuclear activation of NADPH oxidases NOX2 and NOX4 directly contribute to the induction of apoptosis. Previous studies on endothelial cells have shown that p47phox is a crucial subunit in the active endothelial NOX complex by other stimuli, such as high glucose [55], angiotensin [56], and phorbol ester or TNF-α [57]. Interestingly, although evidence shows that NOX4 activity, unlike NOX1 and NOX2, does not depend on p47phox [58], we have found colocalization of p47phox with NOX4 in the peri-nuclear region.
Tyagi et al. demonstrated that incubation with 10–100 μM Hcy for up to 24 h resulted in oxidative stress in cardiac microvascular endothelial cells [59]. These investigators measured intracellular ROS production and correlated it to an increase in NOX1 mRNA expression. However, at that time, they did not study other NOX proteins, nor analyze the intracellular localization of NOX1 and ROS production. Furthermore, under their experimental conditions, Hcy did not cause apoptosis. In our experiments, neither a significant increase in NOX1 expression at protein level was found, nor did cytoplasmic NOX1 expression co-localize with nitrotyrosine, even at the higher concentrations of Hcy. In line with this, Petry et al. [60] demonstrated that NOX2 and NOX4, but not NOX1, contributed to ROS production by and proliferation of endothelial cells, even under basal conditions. A role of NOX4 was also substantiated by Tyagi et al., who found that NOX4 translocated to mitochondria [49] and subsequently that ROS production co-localized with mitochondria and apoptosis [26]. They suggested that Hcy-induced apoptosis in these cells through a mitochondrial mechanism. Indeed, in addition to the dominant (peri-) nuclear accumulation of nitrotyrosine, we also found ROS generation diffusely in the cell after incubation with Hcy (Fig. 4c) and observed that inhibition of mitochondrial ROS production inhibited apoptosis partly.
The accumulation of nitrotyrosine residues not only points to the generation of superoxide, but also to the consumption of nitric oxide [61]. A role of decreased NO availability in Hcy-induced damage has been suggested by other studies [62]. An in vivo study in dogs revealed that Hcy reduces NO bioavailability through generation of superoxide in the heart [63, 64]. Also, in isolated muscle arterioles of HHC rats, xanthine oxidase-derived superoxide scavenges NO, resulting in constriction of the arteriole [43]. Furthermore, Ungvari et al. determined that Hcy increased superoxide which reduced NO availability in endothelium of coronary arteries, resulting in impaired flow [65]. Our current data corroborate with these findings.
An unexpected finding of our study was the nuclear localization of NOX2 and to a minor extent of p47phox. In smooth muscle cells, other authors have shown the nuclear presence of NOX4 [66, 67]. Kuroda et al. [68] also reported NOX4 in the nuclei of endothelial cells. However, in our study, we only observed a perinuclear localization of NOX4 in endothelial cells, similar as Chen et al. [69]. who showed colocalization with the ER marker protein GRP78. Chen et al. suggested that a C-terminal-truncated variant of NOX4 can accumulate in the nucleus. In contrast, the NOX2-specific MoAb-48 recognized NOX in the nucleus of HUVEC. In the light of the foregoing discussion on NOX4, it is of interest to note that the MoAb recognizes the C-terminal part of NOX2. In addition to NOX2, phox47 also was encountered together with nitrosylated proteins, which reflects the presence of NOX activity. As this activity is enhanced after incubation of endothelial cells with Hcy, it would be of interest to further elucidate whether nuclear NOX activity is involved in nuclear changes accompanying apoptosis, or whether it has also other functions such as activation/modulation of transcription factors and cell proliferation [66, 67].
The present study has the limitation that the concentrations of Hcy that we used are relatively high compared to physiological concentrations. However, other studies on endothelial cell apoptosis caused by Hcy also used comparable or even higher concentrations of Hcy up to 10 mM [23–26]. Moreover, the short-term exposure (6 h) to higher Hcy concentrations may reflect a life-long exposure to moderately elevated levels of Hcy, as that occurs in patients. Furthermore, patients suffering from homocystinuria revealed Hcy levels ranging from 200 to 400 μM [70]. Comparing these levels with the concentrations used in our present study, we have tested a threefold higher concentration than that physiologically occurs in these patients, taking into account that only the l-form of Hcy has an effect on our HUVECs and not the d-form [37, 38]. Although the present data suggest a general mechanism in different types of endothelial cells, additional studies on individual NOX proteins in different types of endothelial cells are needed to verify whether our observation is general or whether artery, vein, and specific microvascular endothelial cells display specific responses.
In conclusion, we have shown that Hcy induces apoptosis of endothelial cells, and accumulation of nuclear NOX2 and peri-nuclear NOX4 accompanied by increased amounts of nitrotyrosine. These NOXs provide a potential source for ROS production in Hcy-induced apoptosis of endothelial cells.
References
Carson, N. A., & Neill, D. W. (1962). Metabolic abnormalities detected in a survey of mentally backward individuals in Northern Ireland. Archives of Disease in Childhood, 37, 505–513.
Gerritsen, T., Vaughn, J. G., & Waisman, H. A. (1962). The identification of homocystine in the urine. Biochemical and Biophysical Research Communications, 9, 493–496.
Mudd, S. H., Finkelstein, J. D., Irreverre, F., & Laster, L. (1964). Homocystinuria: an enzymatic defect. Science, 143, 1443–1445.
Gibson, J. B., Carson, N. A., & Neill, D. W. (1964). Pathological findings in homocystinuria. Journal of Clinical Pathology, 17, 427–437.
Frosst, P., Blom, H. J., Milos, R., et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nature Genetics, 10, 111–113.
Symons, J. D., Mullick, A. E., Ensunsa, J. L., Ma, A. A., & Rutledge, J. C. (2002). Hyperhomocysteinemia evoked by folate depletion: effects on coronary and carotid arterial function. Arteriosclerosis Thrombosis and Vascular Biology, 22, 772–780.
McCully, K. S. (1969). Vascular pathology of homocystinemia: implications for the pathogenesis of arteriosclerosis. The American Journal of Pathology, 56, 111–128.
Clarke, R., Daly, L., Robinson, K., et al. (1991). Hyperhomocysteinemia: an independent risk factor for vascular disease. The New England Journal of Medicine, 324, 1149–1155.
Eberhardt, R. T., Forgione, M. A., Cap, A., et al. (2000). Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. The Journal of Clinical Investigation, 106, 483–491.
Lawrence de Koning, A. B., Werstuck, G. H., Zhou, J., & Austin, R. C. (2003). Hyperhomocysteinemia and its role in the development of atherosclerosis. Clinical Biochemistry, 36, 431–441.
Castro, R., Rivera, I., Martins, C., et al. (2005). Intracellular S-adenosylhomocysteine increased levels are associated with DNA hypomethylation in HUVEC. The Journal of Molecular Medicine, 83, 831–836.
Lonn, E., Yusuf, S., Arnold, M. J., et al. (2006). Homocysteine lowering with folic acid and B vitamins in vascular disease. The New England Journal of Medicine, 354, 1567–1577.
Bonaa, K. H., Njolstad, I., Ueland, P. M., et al. (2006). Homocysteine lowering and cardiovascular events after acute myocardial infarction. The New England Journal of Medicine, 354, 1578–1588.
Loscalzo, J. (2006). Homocysteine trials–clear outcomes for complex reasons. The New England Journal of Medicine, 354, 1629–1632.
Chambers, J. C., McGregor, A., Jean-Marie, J., Obeid, O. A., & Kooner, J. S. (1999). Demonstration of rapid onset vascular endothelial dysfunction after hyperhomocysteinemia: an effect reversible with vitamin C therapy. Circulation, 99, 1156–1160.
Bellamy, M. F., & McDowell, I. F. (1997). Putative mechanisms for vascular damage by homocysteine. Journal of Inherited Metabolic Disease, 20, 307–315.
Woo, K. S., Chook, P., Lolin, Y. I., et al. (1997). Hyperhomocyst(e)inemia is a risk factor for arterial endothelial dysfunction in humans. Circulation, 96, 2542–2544.
Lentz, S. R., Sobey, C. G., Piegors, D. J., et al. (1996). Vascular dysfunction in monkeys with diet-induced hyperhomocyst(e)inemia. The Journal of Clinical Investigation, 98, 24–29.
De Vriese, A. S., Blom, H. J., Heil, S. G., et al. (2004). Endothelium-derived hyperpolarizing factor-mediated renal vasodilatory response is impaired during acute and chronic hyperhomocysteinemia. Circulation, 109, 2331–2336.
Harker, L. A., Ross, R., Slichter, S. J., & Scott, C. R. (1976). Homocystine-induced arteriosclerosis. The role of endothelial cell injury and platelet response in its genesis. The Journal of Clinical Investigation, 58, 731–741.
Wilson, K. M., McCaw, R. B., Leo, L., et al. (2007). Prothrombotic effects of hyperhomocysteinemia and hypercholesterolemia in ApoE-deficient mice. Arteriosclerosis Thrombosis and Vascular Biology, 27, 233–240.
Heydrick, S. J., Weiss, N., Thomas, S. R., et al. (2004). l-Homocysteine and l-homocystine stereospecifically induce endothelial nitric oxide synthase-dependent lipid peroxidation in endothelial cells. Free Radical Biology and Medicine, 36, 632–640.
Kerkeni, M., Tnani, M., Chuniaud, L., Miled, A., Maaroufi, K., & Trivin, F. (2006). Comparative study on in vitro effects of homocysteine thiolactone and homocysteine on HUVEC cells: evidence for a stronger proapoptotic and pro-inflammative homocysteine thiolactone. Molecular and Cellular Biochemistry, 291, 119–126.
Suhara, T., Fukuo, K., Yasuda, O., et al. (2004). Homocysteine enhances endothelial apoptosis via upregulation of Fas-mediated pathways. Hypertension, 43, 1208–1213.
Zhang, C., Cai, Y., Adachi, M. T., et al. (2001). Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. The Journal of Biological Chemistry, 276, 35867–35874.
Tyagi, N., Ovechkin, A. V., Lominadze, D., Moshal, K. S., & Tyagi, S. C. (2006). Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. Journal of Cellular Biochemistry, 98, 1150–1162.
Dong, F., Zhang, X., Li, S. Y., et al. (2005). Possible involvement of NADPH oxidase and JNK in homocysteine-induced oxidative stress and apoptosis in human umbilical vein endothelial cells. Cardiovascular Toxicology, 5, 9–20.
Meischl, C., Krijnen, P. A., Sipkens, J. A., et al. (2006). Ischemia induces nuclear NOX2 expression in cardiomyocytes and subsequently activates apoptosis. Apoptosis, 11, 913–921.
Sipkens, J. A., Krijnen, P. A., Meischl, C., et al. (2007). Homocysteine affects cardiomyocyte viability: concentration-dependent effects on reversible flip-flop, apoptosis and necrosis. Apoptosis, 12(8), 1407–1418.
Verhoeven, A. J., Bolscher, B. G., Meerhof, L. J., et al. (1989). Characterization of two monoclonal antibodies against cytochrome b558 of human neutrophils. Blood, 73, 1686–1694.
Burritt, J. B., Fritel, G. N., Dahan, I., Pick, E., Roos, D., & Jesaitis, A. J. (2000). Epitope identification for human neutrophil flavocytochrome b monoclonals 48 and 449. European Journal of Haematology, 65, 407–413.
Krijnen, P. A., Meischl, C., Hack, C. E., et al. (2003). Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. Journal of Clinical Pathology, 56, 194–199.
Nakazawa, H., Fukuyama, N., Takizawa, S., Tsuji, C., Yoshitake, M., & Ishida, H. (2000). Nitrotyrosine formation and its role in various pathological conditions. Free Radical Research, 33, 771–784.
Ye, Y. Z., Strong, M., Huang, Z. Q., & Beckman, J. S. (1996). Antibodies that recognize nitrotyrosine. Methods in Enzymology, 269, 201–209.
Zheng, L., Nukuna, B., Brennan, M. L., et al. (2004). Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. The Journal of Clinical Investigation, 114, 529–541.
Mercie, P., Garnier, O., Lascoste, L., et al. (2000). Homocysteine-thiolactone induces caspase-independent vascular endothelial cell death with apoptotic features. Apoptosis, 5, 403–411.
Poddar, R., Sivasubramanian, N., DiBello, P. M., Robinson, K., & Jacobsen, D. W. (2001). Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation, 103, 2717–2723.
Weiss, N. (2005). Mechanisms of increased vascular oxidant stress in hyperhomocys-teinemia and its impact on endothelial function. Current Drug Metabolism, 6, 27–36.
Ubbink, J. B., Hayward Vermaak, W. J., & Bissbort, S. (1991). Rapid high-performance liquid chromatographic assay for total homocysteine levels in human serum. Journal of Chromatography, 565, 441–446.
Struys, E. A., Jansen, E. E., de Meer, K., & Jakobs, C. (2000). Determination of S-adenosylmethionine and S-adenosylhomocysteine in plasma and cerebrospinal fluid by stable-isotope dilution tandem mass spectrometry. Clinical Chemistry, 46, 1650–1656.
Afri, M., Frimer, A. A., & Cohen, Y. (2004). Active oxygen chemistry within the liposomal bilayer. Part IV: locating 2′, 7′-dichlorofluorescein (DCF), 2′, 7′-dichlorodihydrofluorescein (DCFH) and 2′, 7′- dichlorodihydrofluorescein diacetate (DCFH-DA) in the lipid bilayer. Chemistry and Physics of Lipids, 131, 123–133.
Juffermans, L. J., Dijkmans, P. A., Musters, R. J., Visser, C. A., & Kamp, O. (2006). Transient permeabilization of cell membranes by ultrasound-exposed microbubbles is related to formation of hydrogen peroxide. American Journal of Physiology Heart and Circulatory Physiology, 291, H1595–H1601.
Jacobi, J., Kristal, B., Chezar, J., Shaul, S. M., & Sela, S. (2005). Exogenous superoxide mediates pro- oxidative, proinflammatory, and procoagulatory changes in primary endothelial cell cultures. Free Radical Biology and Medicine, 39, 1238–1248.
Bao, X. M., Wu, C. F., & Lu, G. P. (2009). Atorvastatin attenuates homocysteine-induced apoptosis in human umbilical vein endothelial cells via inhibiting NADPH oxidase-related oxidative stress- triggered p38MAPK signaling. Acta Pharmacologica Sinica, 30, 1392–1398.
Xu, Z., Lu, G., & Wu, F. (2009). Simvastatin suppresses homocysteine-induced apoptosis in endothelial cells: roles of caspase-3, cIAP-1 and cIAP-2. Hypertension Research, 32, 375–380.
Lee, S. J., Kim, K. M., Namkoong, S., Kim, C. K., Kang, Y. C., Lee, H., et al. (2005). Nitric oxide inhibition of homocysteine-induced human endothelial cell apoptosis by down-regulation of p53-dependent NOXa expression through the formation of S-nitrosohomocysteine. The Journal of Biological Chemistry, 280, 5781–5788.
Perez-de-Arce, K., Foncea, R., & Leighton, F. (2005). Reactive oxygen species mediates homocysteine- induced mitochondrial biogenesis in human endothelial cells: modulation by antioxidants. Biochemical and Biophysical Research Communications, 338, 1103–1109.
Tsen, C. M., Hsieh, C. C., Yen, C. H., & Lau, Y. T. (2003). Homocysteine altered ROS generation and NO accumulation in endothelial cells. Chinese Journal of Physiology, 46, 129–136.
Tyagi, N., Moshal, K. S., Sen, U., Lominadze, D., Ovechkin, A. V., & Tyagi, S. C. (2006). Ciglitazone ameliorates homocysteine-mediated mitochondrial translocation and matrix metalloproteinase-9 activation in endothelial cells by inducing peroxisome proliferator activated receptor-gamma activity. Cellular and Molecular Biology (Noisy-le-Grand, France), 52, 21–27.
Edirimanne, V. E., Woo, C. W., Siow, Y. L., Pierce, G. N., Xie, J. Y., & O, K. (2007). Homocysteine stimulates NADPH oxidase-mediated superoxide production leading to endothelial dysfunction in rats. Canadian Journal of Physiology and Pharmacology, 85, 1047–1236.
Tyagi, N., Moshal, K. S., Sen, U., Vacek, T. P., Kumar, M., Hughes, W. M., Jr, et al. (2009). H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxidants and Redox Signaling, 11, 25–33.
Li, N., Yi, F. X., Rute, E., Zhang, D. X., Slocum, G. R., & Zou, A. P. (2002). Effects of homocysteine on intracellular nitric oxide and superoxide levels in the renal arterial endothelium. American Journal of Physiology Heart and Circulatory Physiology, 283, H1237–H1243.
Moshal, K. S., Singh, M., Sen, U., Rosenberger, D. S., Henderson, B., Tyagi, N., et al. (2006). Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MP-9 in MVEC. American Journal of Physiology Heart and Circulatory Physiology, 291, H2825–H2835.
Cai, Y., Zhang, C., Nawa, T., et al. (2000). Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood, 96, 2140–2148.
Quagliaro, L., Piconi, L., Assaloni, R., Martinelli, L., Motz, E., & Ceriello, A. (2003). Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes, 52, 2795–2804.
Li, J. M., & Shah, A. M. (2003). Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. The Journal of Biological Chemistry, 278, 12094–12100.
Li, J. M., Mullen, A. M., Yun, S., et al. (2002). Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-alpha. Circulation Research, 90, 143–150.
Martyn, K. D., Frederick, L. M., von Loehneysen, K., Dinauer, M. C., & Knaus, U. G. (2006). Functional analysis of NOX4 reveals unique characteristics compared to other NADPH oxidases. Cellular Signalling, 18, 69–82.
Tyagi, N., Sedoris, K. C., Steed, M., Ovechkin, A. V., Moshal, K. S., & Tyagi, S. C. (2005). Mechanisms of homocysteine-induced oxidative stress. American Journal of Physiology Heart and Circulatory Physiology, 289, H2649–H2656.
Petry, A., Djordjevic, T., Weitnauer, M., Kietzmann, T., Hess, J., & Gorlach, A. (2006). NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxidants and Redox Signaling, 8, 1473–1484.
Pacher, P., Beckman, J. S., & Liaudet, L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiological Reviews, 87, 315–424.
Loscalzo, J. (1996). The oxidant stress of hyperhomocyst(e)inemia. The Journal of Clinical Investigation, 98, 5–7.
Suematsu, N., Ojaimi, C., Kinugawa, S., et al. (2007). Hyperhomocysteinemia alters cardiac substrate metabolism by impairing nitric oxide bioavailability through oxidative stress. Circulation, 115, 255–262.
Bagi, Z., Ungvari, Z., & Koller, A. (2002). Xanthine oxidase-derived reactive oxygen species convert flow- induced arteriolar dilation to constriction in hyperhomocysteinemia: possible role of peroxynitrite. Arteriosclerosis Thrombosis and Vascular Biology, 22, 28–33.
Ungvari, Z., Csiszar, A., Bagi, Z., & Koller, A. (2002). Impaired nitric oxide-mediated flow-induced coronary dilation in hyperhomocysteinemia: morphological and functional evidence for increased peroxynitrite formation. The American Journal of Pathology, 161, 145–153.
Hilenski, L. L., Clempus, R. E., Quinn, M. T., Lambeth, J. D., & Griendling, K. K. (2004). Distinct subcellular localizations of NOX1 and NOX4 in vascular smooth muscle cells. Arteriosclerosis Thrombosis and Vascular Biology, 24, 677–683.
Lyle, A. N., Deshpande, N. N., Taniyama, Y., et al. (2009). Poldip2, a novel regulator of NOX4 and cytoskeletal integrity in vascular smooth muscle cells. Circulation Research, 105, 249–259.
Kuroda, J., Nakagawa, K., Yamasaki, T., et al. (2005). The superoxide-producing NAD(P)H oxidase NOX4 in the nucleus of human vascular endothelial cells. Genes to Cells, 10, 1139–1151.
Chen, K., Kirber, M. T., Xiao, H., Yang, Y., & Keaney, J. F., Jr. (2008). Regulation of ROS signal transduction by NADPH oxidase 4 localization. The Journal of Cell Biology, 181, 1129–1139.
Tangerman, A., Wilcken, B., Levy, H. L., Boers, G. H., & Mudd, S. H. (2000). Methionine transamination in patients with homocystinuria due to cystathionine beta-synthase deficiency. Metabolism, 49, 1071–1077.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sipkens, J.A., Hahn, N., van den Brand, C.S. et al. Homocysteine-Induced Apoptosis in Endothelial Cells Coincides With Nuclear NOX2 and Peri-nuclear NOX4 Activity. Cell Biochem Biophys 67, 341–352 (2013). https://doi.org/10.1007/s12013-011-9297-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-011-9297-y