
Abstract
Sclerosing bone dysplasias are a group of rare, monogenic disorders characterized by increased bone density resulting from the disturbance in the fragile equilibrium between bone formation and resorption. Over the last decade, major contributions have been made toward better understanding of the pathogenesis of these conditions. These studies provided us with important insights into the bone biology and yielded the identification of numerous drug targets for the prevention and treatment of osteoporosis. Here, we review this heterogeneous group of disorders focusing on their utility in the development of novel osteoporosis therapies.

Similar content being viewed by others

References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377(9773):1276–87.
Ferrari S. Human genetics of osteoporosis. Best Pract Res Clin Endocrinol Metab. 2008;22(5):723–35.
Kornak U et al. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9(13):2059–63.
Sobacchi C et al. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9(9):522–36. Comprehensive and up-to-date review on the heterogeneous group of Osteopetroses.
Kornak U et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104(2):205–15.
Chalhoub N et al. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med. 2003;9:399–406.
Sly WS et al. Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N Engl J Med. 1985;313:139–45.
Guerrini MM et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. 2008;83(1):64–76.
Sobacchi C et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. 2007;39(8):960–2.
Smahi A et al. The NF-kappaB signaling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11(20):2371–5.
Van Wesenbeeck L et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J Clin Invest. 2007;117(4):919–30.
Aker M et al. An SNX10 mutation causes malignant osteopetrosis of infancy. J Med Genet. 2012;49(4):221–6.
Cleiren E et al. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet. 2001;10(25):2861–7.
Bollerslev J et al. Autosomal dominant osteopetrosis revisited: lessons from recent studies. Eur J Endocrinol. 2013;169(2):R39–57.
Gelb BD et al. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273(5279):1236–8.
Little RD et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70(1):11–9.
Boyden LM et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346(20):1513–21.
Ai M et al. Clinical and molecular findings in osteoporosis-pseudoglioma syndrome. Am J Hum Genet. 2005;77(5):741–53.
Balemans W et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–43.
Brunkow ME et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68(3):577–89.
Leupin O et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–500.
Balemans W et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39(2):91–7.
Kim SJ et al. Identification of signal peptide domain SOST mutations in autosomal dominant craniodiaphyseal dysplasia. Hum Genet. 2011;129(5):497–502.
Jenkins ZA et al. Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet. 2009;41(1):95–100.
Nurnberg P et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, results in craniometaphyseal dysplasia. Nat Genet. 2001;28(1):37–41.
Reichenberger E et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001;68(6):1321–6.
Hu Y et al. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia. PLoS One. 2013;8(8):e73576.
Janssens K et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26(3):273–5.
Kinoshita A et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000;26(1):19–20.
Hellemans J et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet. 2004;36(11):1213–8.
Mumm S et al. Deactivating germline mutations in LEMD3 cause osteopoikilosis and Buschke-Ollendorff syndrome, but not sporadic melorheostosis. J Bone Miner Res. 2007;22(2):243–50.
Ababneh FK et al. Hereditary deletion of the entire FAM20C gene in a patient with Raine syndrome. Am J Med Genet A. 2013;161A(12):3155–60.
Wang X et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012;8(5):e1002708.
Singer FR, Mills BG. Evidence for a viral etiology of Paget's disease of bone. Clin Orthop Relat Res. 1983;178:245–51.
Laurin N et al. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70(6):1582–8.
Hocking LJ et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum Mol Genet. 2002;11(22):2735–9.
Johnson JO et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–64.
Whyte MP et al. Osteoprotegerin deficiency and juvenile Paget's disease. N Engl J Med. 2002;347(3):175–84.
Hughes AE et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24(1):45–8.
Whyte MP, Hughes AE. Expansile skeletal hyperphosphatasia is caused by a 15-base pair tandem duplication in TNFRSF11A encoding RANK and is allelic to familial expansile osteolysis. J Bone Miner Res. 2002;17(1):26–9.
Whyte MP et al. Expansile skeletal hyperphosphatasia: a new familial metabolic bone disease. J Bone Miner Res. 2000;15(12):2330–44.
Boudin E et al. No mutations in the serotonin related TPH1 and HTR1B genes in patients with monogenic sclerosing bone disorders. Bone. 2013;55(1):52–6.
Boudin E et al. Mutations in sFRP1 or sFRP4 are not a common cause of craniotubular hyperostosis. Bone. 2013;52(1):292–5.
Pangrazio A et al. Exome sequencing identifies CTSK mutations in patients originally diagnosed as intermediate osteopetrosis. Bone. 2014;59:122–6.
Lazarus S, Zankl A, Duncan EL. Next-generation sequencing: a frameshift in skeletal dysplasia gene discovery. Osteoporos Int. 2014;25(2):407–22. Reviews the insights into Sclerosing Bone Dysplasias provided by novel sequencing techniques.
Borra VM et al. Localization of the gene for hyperostosis cranialis interna to chromosome 8p21 with analysis of three candidate genes. Calcif Tissue Int. 2013;93(1):93–100.
Borra VM et al. Localization of the gene for X-linked calvarial hyperostosis to chromosome Xq27.3-Xqter. Bone. 2014;58:67–71.
Pagon RA, Beckwith JB, Ward BH. Calvarial hyperostosis: a benign X-linked recessive disorder. Clin Genet. 1986;29(1):73–8.
Zhang Y et al. Novel and recurrent germline LEMD3 mutations causing Buschke-Ollendorff syndrome and osteopoikilosis but not isolated melorheostosis. Clin Genet. 2009;75(6):556–61.
Kasapkara CS et al. An extremely rare case: osteosclerotic metaphyseal dysplasia. Genet Couns. 2013;24(1):69–74.
Yao AL, Camacho PM. Osteomesopyknosis: a case report and review of sclerosing bone disorders. Endocr Pract. 2014:1–14. doi:10.4158/EP13352.CR.
Das S, Crockett JC. Osteoporosis - a current view of pharmacologic prevention and treatment. Drug Des Devel Ther. 2013;7:435–48.
Leder BZ, et al. Two years of denosumab and teriparatide administration in postmenopausal women with osteoporosis (The DATA Extension Study): a randomized controlled trial. J Clin Endocrinol Metab. 2014;99(5):1694–700.
Schaller S et al. The chloride channel inhibitor NS3736 [corrected] prevents bone resorption in ovariectomized rats without changing bone formation. J Bone Miner Res. 2004;19(7):1144–53.
Qin A et al. V-ATPases in osteoclasts: structure, function and potential inhibitors of bone resorption. Int J Biochem Cell Biol. 2012;44(9):1422–35.
Saftig P et al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A. 1998;95(23):13453–8.
Duong LT. Therapeutic inhibition of cathepsin K-reducing bone resorption while maintaining bone formation. Bonekey Reports, 20. doi:10.1038/bonekey.2012.67.
Langdahl B et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: 5 years of continued therapy in a phase 2 study. J Bone Miner Res. 2012;27(11):2251–8.
Sims NA, Ng KW. Implications of osteoblast-osteoclast interactions in the management of osteoporosis by antiresorptive agents denosumab and odanacatib. Curr Osteoporos Rep. 2014;12(1):98–106.
Chen M et al. Emerging therapeutic targets for osteoporosis treatment. Expert Opin Ther Target. 2014;18(7):817–831.
Ng KW, Martin TJ. New therapeutics for osteoporosis. Curr Opin Pharmacol. 2014;16C:58–63.
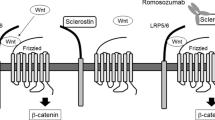
Boudin E et al. The role of extracellular modulators of canonical Wnt signaling in bone metabolism and diseases. Semin Arthritis Rheum. 2013;43(2):220–40.
McClung MR et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412–20. The most recent report on the clinical trials of the most promising osteoporosis treatment in development.
Kedlaya R et al. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med. 2013;5(211):211ra158.
Sinder BP et al. Sclerostin antibody improves skeletal parameters in a Brtl/+mouse model of osteogenesis imperfecta. J Bone Miner Res. 2013;28(1):73–80. Supports the therapeutic effect of sclerostin antibody in murine model of Osteogenesis Imperfecta. Demonstrates the diversity of this therapeutic agent.
Chang MK et al. Reversing LRP5-dependent osteoporosis and SOST deficiency-induced sclerosing bone disorders by altering WNT signaling activity. J Bone Miner Res. 2014;29(1):29–42. Comprehensive studies providing insights into the biology of sclerostin inhibition.
van Lierop AH et al. Serum Dickkopf 1 levels in sclerostin deficiency. J Clin Endocrinol Metab. 2014;99(2):E252–6.
Morvan F et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21(6):934–45.
Moore WJ et al. Modulation of Wnt signaling through inhibition of secreted frizzled-related protein I (sFRP-1) with N-substituted piperidinyl diphenylsulfonyl sulfonamides: part II. Bioorg Med Chem. 2010;18(1):190–201.
Moore WJ et al. Modulation of Wnt signaling through inhibition of secreted frizzled-related protein I (sFRP-1) with N-substituted piperidinyl diphenylsulfonyl sulfonamides. J Med Chem. 2009;52(1):105–16.
Acknowledgments
Research relevant for this review was supported by a grant from the University of Antwerp (TOP-BOF) and 2 research grants (G.0065.10N and G.0197.12N) from the Fonds Wetenschappelijk Onderzoek-Vlaanderen (FWO) to W. Van Hul. I. Fijałkowski holds a pre-doctoral specialization scholarship from the “Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen)”. E. Boudin holds a post-doctoral fellowship of the FWO (Fund for Scientific Research) Vlaanderen.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
I. Fijalkowski, E. Boudin, G. Mortier, and W. Van Hul declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by the authors involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fijalkowski, I., Boudin, E., Mortier, G. et al. Sclerosing Bone Dysplasias: Leads Toward Novel Osteoporosis Treatments. Curr Osteoporos Rep 12, 243–251 (2014). https://doi.org/10.1007/s11914-014-0220-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-014-0220-5
Keywords
- Sclerosing bone dysplasia
- Osteopetrosis
- Pycnodysostosis
- High bone mass phenotype
- Sclerosteosis
- Van Buchem disease
- Craniodiaphyseal dysplasia
- Craniometaphyseal dysplasia
- Camurati-Engelmann disease
- Osteopoikilosis
- Raine syndrome
- Paget’s disease of bone
- Ostaoectasia with hyperphosphatasia,
- Familial expansile osteolysis
- Expansile skeletal hyperphosphatasia
- Osteoporosis
- Treatment