
Abstract
Axonal demyelination is a consistent pathological sequel to chronic brain and spinal cord injuries and disorders that slows or disrupts impulse conduction, causing further functional loss. Since oligodendroglial progenitors are present in the demyelinated areas, failure of remyelination may be due to lack of sufficient proliferation and differentiation of oligodendroglial progenitors. Guanosine stimulates proliferation and differentiation of many types of cells in vitro and exerts neuroprotective effects in the central nervous system (CNS). Five weeks after chronic traumatic spinal cord injury (SCI), when there is no ongoing recovery of function, intraperitoneal administration of guanosine daily for 2 weeks enhanced functional improvement correlated with the increase in myelination in the injured cord. Emphasis was placed on analysis of oligodendrocytes and NG2-positive (NG2+) cells, an endogenous cell population that may be involved in oligodendrocyte replacement. There was an increase in cell proliferation (measured by bromodeoxyuridine staining) that was attributable to an intensification in progenitor cells (NG2+ cells) associated with an increase in mature oligodendrocytes (determined by Rip+ staining). The numbers of astroglia increased at all test times after administration of guanosine whereas microglia only increased in the later stages (14 days). Injected guanosine and its breakdown product guanine accumulated in the spinal cords; there was more guanine than guanosine detected. We conclude that functional improvement and remyelination after systemic administration of guanosine is due to the effect of guanosine/guanine on the proliferation of adult progenitor cells and their maturation into myelin-forming cells. This raises the possibility that administration of guanosine may be useful in the treatment of spinal cord injury or demyelinating diseases such as multiple sclerosis where quiescent oligodendroglial progenitors exist in demyelinated plaques.
Similar content being viewed by others


Introduction
Demyelination contributes to functional deficits in central nervous system (CNS) disorders [1–3]. Traumatic spinal cord injury (SCI) induces local inflammation and demyelination in the white matter around the lesion resulting in disrupted axonal conduction [4–7]. Remyelination is critical for recovery, but does not occur spontaneously [8–11]. Spinal cords contain endogenous progenitors that can proliferate and differentiate into mature oligodendroglia and remyelinate axons under certain circumstances [12–14]. After acute SCI in rats, oligodendrocyte precursors proliferate in the first 2 weeks but are “silent” thereafter [15], possibly because the oligodendroglial precursors cannot differentiate into mature oligodendroglia capable of remyelinating axons. Enhancement of proliferation of oligodendrocytes or progenitors may lead to repair and restoration of function after SCI.
Neurotrophic factors restore physiological function by promoting oligodendrocyte proliferation and axonal sprouting [16–19], regulating the proliferation of oligodendrocyte progenitors [3] and modulating the differentiation [20–22] and the maturation of oligodendrocyte precursors into myelin-forming cells [23, 24].
Guanosine has trophic effects on many cell types [25–28], and we have some evidence that guanine acts similarly. For example, it stimulates proliferation of a variety of cells in culture [25, 28] and promotes synthesis and release of several potentially neuroprotective trophic factors from a variety of cells, including neuron growth factor (NGF) from astrocytes as well as basic fibroblast growth factor (bFGF) and transforming growth factor beta (TGF beta) [25, 27]. Extracellular guanosine itself also enhances outgrowth of nerve processes from PC12 cells and from primary cultures of embryonic rat brain neurons [25]. As well, it acts synergistically with NGF to promote outgrowth of neurites [29, 30].
Our preliminary data [31] showed that 35 days after moderate traumatic injury of the spinal cord of rats, when the initial spontaneous incomplete recovery of function had plateaued, systemic administration of guanosine (intraperitoneally 8 mg/kg body weight) for 7 days enhanced locomotor functional recovery that continued to improve throughout the 7-day duration of the treatment and correlated with the increase in myelination. In the present study, we treated rats for up to 14 days to determine whether guanosine-induced functional improvement was due to triggering the endogenous adult progenitors. Meanwhile, we also examined the uptake and tissue distribution of systemically administered guanosine.
Materials and methods
All experiments were performed in compliance with the requirements of the Animals for Research Act of Ontario, Canada and the Guidelines of the Canadian Council on Animal Care, and had been approved by the Animal Research Ethics Board of McMaster University.
Spinal cord injury
Adult female Wistar rats (280–300 g weight, Charles River) were anaesthetized with isoflurane (3–5%): O2 (1 l/min). Buprenorphine (0.03 mg/kg body weight, subcutaneously) was administered prior to surgery for pain relief. Moderate spinal cord injury was induced by crushing exposed cords with modified coverslip forceps [31–33]. The forceps were closed slowly over 2 s and the compression sustained for 15 s. The muscles and fat pad over the lesion were sutured and the skin was closed with stainless steel clips. Post-operatively, the rats were kept quiet and warm [31, 34, 35].
Behavioural assessment and drug administration
Rats were handled daily for 2 weeks pre-operatively to acclimatize them to the experimental procedures and behavioural test. After the spinal injury, locomotor recovery was assessed weekly for 35 days in an open field walking task (OFWT) before treatment. Cagemates (two animals) were placed in the centre of the open field, formed by a child’s circular plastic swimming pool (1.3 m in diameter). They were observed for 5-min periods and scored for general locomotor ability using the Basso-Beattie-Bresnahan (BBB) locomotor rating scale [36, 37]. Rats were rated on a scale of 0 to 21, with 0 being no function and 21 being normal. If the animal stopped moving for a minute, it was placed again in the centre of the open field; otherwise it was left alone for the duration of the 5-min test period.
On day 35 following the injury, rats were randomly assigned to three groups. One group of rats was used for baseline behavioural and histological analysis. The other rats received either daily intraperitoneal (i.p.) injections of 8 mg/kg guanosine or the same volume of saline containing 0.001 N NaOH [31] for 2 weeks. The open field walking testing was performed once a day from days 35 to 48 post-surgery (0 to 14 days post-treatment). Groups of 9–12 rats were killed by perfusion/fixation for immunohistochemical analysis at 1, 3, 7 or 14 days after treatment.
BrdU administration
Bromodeoxyuridine (BrdU) was used to label proliferating cells after treatment. BrdU (50 mg/kg in sterile saline; Sigma, B-5002) was injected intraperitoneally daily for 1, 3, 7 or 14 consecutive days, beginning the day treatment was initiated, thereby reflecting cell proliferation over the entire treatment period.
Tissue processing and immunohistochemistry
On post-operative days 35, 36, 38, 42 or 49 (representing baseline, 1, 3, 7 or 14 days treatment), rats were deeply anaesthetized with sodium pentobarbital (50–60 mg/kg body weight, i.p.) immediately following the final locomotor testing and perfused transcardially with 100 ml 0.1 M phosphate-buffered saline (PBS) containing 0.1% heparin, followed by 300–500 ml of 4% paraformaldehyde (PFA). T9 to L1 segments of the spinal cords were removed and incubated in the same fixative for 2 h at room temperature and then cryoprotected in 30% sucrose PBS solution. A segment of each cord, extending from 5 mm rostral to 5 mm caudal to the lesion site was embedded in medium (Tissue-Tek® O.C.T. compound, Sakura Finetek USA, Inc., Torrance, CA, USA). Serial transverse sections were cut at 15- to 20-μm intervals on a cryostat and mounted onto slides (ColorFrost/Plus; Fisher, Pittsburgh, PA, USA). Every fifth section was stained with the lipophilic dye Luxol fast blue to stain myelin [38]. Adjacent sections were stained immunohistochemically with the various antibodies described below.
For BrdU immunohistochemistry, 6% hydrogen peroxide was used to reduce endogenous peroxidase activity, followed by three 10-min PBS rinses and incubation in 1 N HCl at 37°C for 30 min. After rinsing, a blocking solution [5% bovine serum albumin (BSA) in 0.1% Triton-100] was applied for 1 h; sections were subsequently incubated in rat anti-BrdU (1:200 in 1% BSA in 0.1% Triton-100; Serotec, Raleigh, NC, USA) at 4°C overnight in a humid chamber. Sections were then rinsed and incubated with biotinylated horse anti-rat IgG secondary antibodies. Sections were then incubated with Elite ABC (Vector Laboratories, Burlingame, CA, USA) for 1 h, followed by 3,3′-diaminobenzidine (DAB) substrate kit for peroxidase (Vector Laboratories, Burlingame, CA, USA) for 5–10 min. Sections were dehydrated, coverslipped and examined under a microscope.
To determine the fate of newly born cells after SCI, double-fluorescent immunolabelling was performed, combining BrdU with one of the cell-specific phenotypic markers listed below. For double immunofluorescence, sections were pretreated with HCl as described above and incubated with BrdU antibody mixed with one of the following antibodies in PBS at 4°C overnight in a humid chamber: rabbit anti-MBP (myelin basic protein) polyclonal antibodies (1:50; Chemicon Int., Temecula, CA, USA), rabbit anti-NG2 polyclonal antibodies (chondroitin sulfate proteoglycan for oligodendroglial progenitors; 1:200; Chemicon Int. Temecula, CA, USA), mouse anti-Rip monoclonal antibodies (mature oligodendrocyte-specific marker: Reactive Immunology Protein; 1:200; Chemicon Int., Temecula, CA, USA; [39]), rabbit anti-glial fibrillary acidic protein (GFAP) polyclonal antibodies (1:600; Zymed® Lab-SA System Kit, San Francisco, CA, USA) and mouse anti-microglial-specific marker OX-42 monoclonal antibodies (1:200; CD11b, Research Diagnostics Inc., Flanders, NJ, USA). For Rip and OX-42 monoclonal antibodies single staining, sections were developed with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG. For BrdU and polyclonal antibodies (NG2 or MBP or GFAP) double immunolabelling, sections were developed using a mixture of FITC-conjugated goat anti-rat IgG and rhodamine-conjugated goat anti-rabbit IgG in 1% normal goat serum and 0.25% Triton X-100; 1:200 (Invitrogen, Carlsbad, CA, USA) for 2 h. To examine the contribution of Schwann cells from the peripheral nervous system (PNS) in the remyelination process, sections were immunolabelled with specific markers for Schwann cell myelin P0 (a rabbit polyclonal anti-P0 antibody was kindly donated by Prof. Marie T. Filbin, Department of Biology, CUNY, NY, USA; 1:200) and secondary antibody using rhodamine-conjugated goat anti-rabbit IgG. For detection of apoptotic cells, a terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labelling (TUNEL) stain was performed using the ‘In situ Cell Death Detection Kit, Fluorescein’ (Roche Molecular Biochemicals, Mannheim, Germany), according to the manufacturer’s instructions.
Quantification
To quantify the amount of myelin present at the injury site before and after treatment, the Luxol fast blue-stained section from each cord (n = 7–10 for each group) that contained the greatest lesion area was selected together with the adjacent two caudal and two rostral Luxol blue-stained sections. Luxol fast blue staining was also done on cords of five additional unoperated normal animals for comparative purposes. Digital photographs were taken of the sections. The total area of each section and the area of the fast blue-stained portion were measured using a computerised Bioquant BQ-TCW98 image analysis program by an investigator who was blind to group assignment.
For quantification of cell proliferation, five to seven sections taken from the penumbra of the lesion and spaced about 100 μm apart were analysed for each animal (n = 7–9 animals per group). Cellular proliferation was determined by counting the total number of BrdU+ nuclei in spinal cord cross sections. Low power sections were digitized and manually outlined using an image analysis system. Any cavities present in the sections were excluded from analysis. Positively labelled nuclei were counted automatically. The threshold was set such that only positively labelled profiles of the appropriate size were counted. Data are expressed as cells per section.
The total number of NG2+ progenitor cells and the number of those that proliferated (double-labelled for NG2 and BrdU: NG2+/BrdU+) was determined by manual counting. Cell counting was done conservatively, and a positive profile was counted as a cell only when clearly defined borders could be detected completely around and adjacent to the entire nucleus. Data are expressed as the number of immunostained cells per section.
Mature oligodendrocytes in injured cords were identified with Rip, a specific marker for mature oligodendrocytes that recognises an unknown epitope [39] and has been used successfully in the CNS to label both oligodendrocyte processes and myelin sheaths. The number of mature oligodendrocyte (Rip+ cells) in the tissue sections was determined by manual counting.
Evaluation of exogenous guanosine distribution
A guanosine solution (8 mg/kg) was administered intraperitoneally to rats with spinal cord injury induced 5 weeks previously. Samples of spinal cord were taken from the site of the lesion and immediately above or below the lesion just before the intraperitoneal injection of guanosine and at 7.5, 30 and 60 min after the guanosine injection. There were between three and six animals at each time point. Samples of spinal cord were placed in 0.5 ml of cold 0.4 M perchloric acid solution and immediately homogenized. Samples were then centrifuged (4°C, 10,000 g, 10 min) to remove proteins precipitated by perchloric acid and the supernates containing the soluble molecules were neutralized with KOH solution and centrifuged (4°C, 10,000 g, 5 min). Samples were filtered with 0.2-μm filters (Millipore, Billerica, MA, USA) and stored at −70°C before high-performance liquid chromatography (HPLC) [40] or capillary electrophoresis (CE) analysis.
Tissue concentrations of guanosine and guanine
To evaluate the concentration of guanosine and its principal degradation product, guanine, in spinal cords before and after the intraperitoneal injection of guanosine, either HPLC or CE was used. There was no significant difference in the values obtained by these two methods so the data represent the mean of values obtained by both analyses. HPLC analysis was carried out using an Agilent 1100 series HPLC (Agilent Technologies, Waldbronn, Germany) and an ion-pair technique. Separation was carried out with a reverse-phase analytical column (LiChroCART 125-4 LiChrospher 100 RP-18 5 μm, Merck, Darmstadt, Germany) and the flow rate was 1.5 ml/min. A 15-min linear gradient was applied from 100% buffer A (60 mM KH2PO4 and 5 mM tetrabutylammonium phosphate, pH 6) to 100% buffer B (30% methanol plus 70% buffer A). CE separations were carried out using an HP3DCE capillary electrophoresis system (Agilent Technologies, Waldbronn, Germany) equipped with a diode array detection system. Analysis was performed using a 80.5 cm × 50 μm ID × 375 μm OD uncoated fused silica capillary from Agilent Technologies (Waldbronn, Germany) with an effective length of 72 cm to the detector window. The capillary temperature was kept constant at 37°C and the running buffer was 10 mM borate, pH 9.3. The samples were injected by pressure (50 mb for 40 s) and the separation was conducted at 22 kV. Detection took place at 254 nm and purines were identified by their migration times and UV spectra using the diode array detector (Agilent Technologies, Waldbronn, Germany).
Statistical analysis
Data are expressed as mean ± SEM. Behavioural scores were analysed by the Kruskal-Wallis non-parametric analysis of variance (ANOVA). Post hoc comparisons were made using the Dunnett’s test. Correlation between behavioural and histological outcomes was analysed using regression analysis. Significance was set at p < 0.05.
Results and discussion
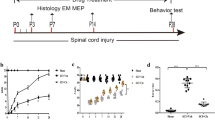
After SCI, motor function in the legs partially recovered over 3–4 weeks, after which there was no further improvement (Fig. 1a). Daily guanosine for 14 days (8 mg/kg intraperitoneally beginning 35 days after injury) produced improvement in locomotor performance over the next 2 weeks; control rats showed no improvement (Fig. 1b). This treatment regimen was associated with significantly increased amounts of myelin in the penumbra of the cord lesions shown by Luxol fast blue staining (Fig. 1c–e). The extent of Luxol fast blue staining in the injured cords correlated with each animal’s open field walking scores (r = 0.72; p < 0.05; Fig. 1f), indicating that guanosine-induced functional recovery may be related to the increase in myelin. Immunostaining for CNS myelin basic protein (MBP; Fig. 1g,h) and a specific marker for Schwann cell myelin, P0 (Fig. 1i,j) showed that Schwann cells from the peripheral nervous system did not enter spinal cord and, therefore, did not contribute to the remyelination.


Systemic treatment with guanosine improves locomotor function and correlates with increased myelination in rats with stable chronic spinal cord injury. a After spinal cord crush, locomotor function was assessed in an open field walking task (OFWT) and scored using a standard scale (“Materials and methods”). No further improvement in locomotor performance occurred after 28 days. b On days 35–48 after injury (days 1 to 14 of treatment in the figure; the first test done immediately prior to the first treatment), animals were randomly divided into two groups. One group received guanosine (▴), the other group received vehicle (♦). The locomotor function in the control group did not change significantly throughout the 14 days of vehicle administration. In contrast, rats that received guanosine showed a marked improvement in locomotor behaviour, which was significantly (p < 0.05) better than vehicle-treated control animals from the 5th day of treatment onward. c, d Luxol fast blue staining of cross sections of spinal cords from guanosine- (d) and vehicle- (c) treated animals at the lesion site 48 days after the initial lesion (after 14 days treatment) shows increased Luxol fast blue-stained material in d compared to the vehicle-treated control group in c. DC dorsal column at the lesion. Scale bar = 50 μm. e The total area of each section and of the Luxol fast blue-stained portion were measured following the protocol in “Materials and methods”. The quantitative results show that the cords from guanosine-treated animals had significantly greater myelinated area (*p < 0.05), as estimated from the Luxol fast blue staining, than cords from vehicle-treated control animals, which in turn had significantly less myelinated area (**p < 0.001) than unoperated animals. f Correlation between behavioural and histological outcomes was analysed using regression analysis. The data show that the Luxol fast blue-stained area in the injured cords from animals treated with guanosine correlated with their behavioural recovery in open field walking scores, correlation coefficient of 0.72, indicating that the recovery of function may be related to the increase in myelin. Myelin immunofluorescent staining of cross sections of spinal cords from vehicle- (g) and guanosine- (h) treated animals at the lesion site 14 days after treatment shows that guanosine-treated cords (h) contained more myelin basic protein (MBP)-positive profiles from central nervous system at the injured site compared to the vehicle-treated control group (g). IS injured site. Whereas, in i (vehicle-treated cord) and j (guanosine-treated cord) immunolabelling with specific markers for Schwann cell myelin P0 indicated that Schwann cells from the peripheral nervous system did not contribute to guanosine-induced remyelination. Scale bar = 50 μm for all
To determine whether existing cells elaborated the myelin or whether it was a product of other cells that proliferated and subsequently elaborated myelin, the number of proliferating cells was determined by counting all BrdU+ nuclei in cross sections of the lesion penumbra at different times after treatment. In the first 3 days after treatment, cell proliferation at the injury site in both guanosine-treated and control animals was not different from baseline (immediately before treatment; Fig. 2c), although at 3 days there was a trend toward more BrdU+ cells in guanosine-treated animals than controls (112 ± 25 vs 79 ± 16). At 7 and 14 days after guanosine treatment, the number of BrdU+ cells increased significantly; there were 382 ± 42 BrdU+ cells per section at 7 days and 477 ± 49 at 14 days compared with only 88 ± 20 at 7 days and 95 ± 22 at 14 days in controls (p < 0.05; Fig. 2a–c). There were few dividing cells beyond the lesion margins.


Quantitative assessment of immunostaining shows that systemic treatment with guanosine stimulates endogenous oligodendroglial progenitors (NG2+ cells) to proliferate and mature in rats with stable chronic spinal cord injury. a, b Bromodeoxyuridine (BrdU) was used to label proliferating cells after treatment. BrdU immunostaining in cross sections from vehicle- (a) and guanosine- (b) treated animals at the lesion 14 days after treatment shows increased BrdU+ nuclei in b compared to the vehicle-treated control group in a. Scale bar = 50 μm. c Quantitative analysis shows that 7 or 14 days after guanosine treatment there was a significant increase in the number of BrdU+ cells compared with vehicle-treated controls. d, e Demonstrate examples of double-fluorescent immunostaining using antibodies against BrdU (in green) and a marker (NG2) for oligodendroglial progenitors (in red) in cross sections from vehicle- (d) and guanosine- (e) treated animals at the lesion 14 days after treatment. Scale bar = 50 μm. f–h Show quantification of proliferating progenitors (NG2+/BrdU+ double-labelled cells; f) and total number of NG2 cells (g). Data indicate that after 7 or 14 days administration guanosine significantly stimulates proliferation of oligodendroglial progenitors (f, g). Concurrently, guanosine also significantly increases the number of mature oligodendroglia shown in i and j compared to vehicle-treated controls (h, j) using fluorescent immunostaining with a specific marker (Rip) to label the mature oligodendroglia. Scale bar = 50 μm
Oligodendrocytes that survive the demyelinating insult are not thought to be able to contribute to remyelination [41, 42]. Instead, endogenous oligodendrocyte progenitor cells local to the lesion are believed to be the source of new myelinating cells [43]. Previous studies have illustrated that NG2+ cells are the major cycling cell within the resting adult CNS [14, 44] and express many characteristics of oligodendroglial lineage cells, not least their ability to differentiate into oligodendrocytes when isolated into culture. Their role in demyelination and remyelination has been extensively studied and there is evidence that they are the cells responsible for myelin repair [43, 45–48]. To determine whether the proliferating cells were related to NG2+ oligodendroglial progenitor cells, we counted the total number of NG2+ progenitor cells and the number that had proliferated (NG2+/BrdU+) from 2 mm rostral to 2 mm caudal to the lesion site (Fig. 2d,e). There was minimal turnover in the NG2+ cells in the spinal cord during the first 3 days in both guanosine-treated and control animals (13 ± 5 in the guanosine group vs 9 ± 4 cells per section in controls; Fig. 2f). By 7 days, guanosine significantly stimulated progenitor proliferation (85 ± 18 vs 13 ± 4 NG2+/BrdU+ cells/section; Fig. 2f); these cells were observed throughout the penumbra with the majority localised in the dorsal columns. Enhanced proliferation of the NG2+ progenitors after guanosine persisted, and by 14 days, double-labelled progenitors in the guanosine-treated group were eightfold more than in controls (108 ± 12 vs 13 ± 5 NG2+/BrdU+ cells/section; Fig. 2f).
The total number of NG2+ cells was also determined. During the first 3 days, there was no significant difference in the numbers of NG2+ cells in cords at baseline after vehicle treatment or guanosine treatment (Fig. 2g). By 7 days, guanosine treatment increased the total NG2+ cells from 96 ± 22 cells/section at baseline to 266 ± 38 cells/section—more than 2 times higher than in the control group (110 ± 24 /section; Fig. 2g). By 14 days, the numbers of double-labelled progenitors in the cords of guanosine-treated animals were threefold higher than controls (347 ± 36 NG2+ cells/section for guanosine-treated animals; 115 ± 24 cells/section for controls; Fig. 2g).
Mature oligodendrocytes were identified with Rip+ staining (Fig. 2h,i). Guanosine had no effect on mature oligodendroglia 3 days after treatment; at 7 and 14 days, there was a significant increase in Rip+ cells in the penumbra of the cords of animals that had received guanosine compared to controls (7 days: 468 ± 40 vs 205 ± 25; and at 14 days: 492 ± 43 vs 219 ± 23 per section, p < 0.01; Fig. 2j).
Guanosine treatment stimulated astrocyte proliferation in the penumbra as early as 3 days and throughout the 14 days of treatment (Fig. 3a,b) and was detected by antibodies to glial fibrillary acidic protein (GFAP). Astrocytes may stimulate oligodendroglial progenitor proliferation or induce their migration [49]. Guanosine did not alter the numbers of OX-42+ microglia/macrophages until 14 days after treatment. At day 14, guanosine significantly increased OX-42+ cells compared to vehicle treatment (Fig. 3c–e; p < 0.05).

Cross sections of the cords at the injury site staining with GFAP and OX-42 from guanosine-treated and vehicle-treated rats 14 days after treatment following chronic spinal cord injury. a, b Demonstrate examples of GFAP-positive astrocytes (in red) in cross sections from vehicle- (a) and guanosine- (b) treated animals at the injury site 14 days after treatment. c, d Show OX-42-immunofluorescent staining of cross sections of spinal cords from vehicle- (c) and guanosine- (d) treated animals at the lesion site 14 days after treatment. Quantification showed more OX-42-immunolabelled cells in cords of guanosine-treated rats (d, e) compared to cords of vehicle-treated rats (c, e; p < 0.05). Scale bar = 50 μm for all
Guanosine inhibits apoptosis due to a number of stimuli [50–52]. After stroke and spinal cord injury, intrinsic stem cells and progenitors proliferate and differentiate but many die through apoptosis [15, 53–55]. In these experiments, guanosine did not prevent cell death (measured by TUNEL) (Fig. 4), indicating that the protective effects of guanosine do not result from a reduction in precursor cell apoptosis.

Guanosine treatment did not affect apoptosis in the cords with chronic, stable spinal cord injury. For detection of apoptotic cells, a terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labelling (TUNEL) stain was performed using the ‘In situ Cell Death Detection Kit, Fluorescein’. No obvious difference in number of TUNEL-positive cells was observed between vehicle- (a) or guanosine- (b) treated spinal cord sections. Scale bar = 50 μm for all
Before the intraperitoneal injection of guanosine, there were detectable amounts of guanosine and its metabolic product guanine in the spinal cords of injured rats (time 0, Fig. 5a,b). There were no significant differences in guanosine or guanine in the samples of the spinal cords above, at or below the lesion. After guanosine injection, the amount of both guanosine and guanine increased, reaching a maximum by 30 min; the amounts of guanosine and guanine were about twofold and fivefold higher than those detected before the injection (Fig. 5a,b). As with the basal levels (time 0), there was no difference in the amounts of guanosine above, at or below the lesion. Interestingly, guanine levels were always higher than those of guanosine at each of the times tested after injection (Fig. 5a,b).

Guanosine (a) and guanine (b) in spinal cord samples of rats with chronic spinal cord injury before and after intraperitoneal injection of guanosine. Guanosine (8 mg/kg) was administered intraperitoneally to rats 5 weeks after the spinal cord injury. Before or at the indicated time points after the injection, samples of spinal cord from 3–6 animals were taken from the site of the lesion and immediately above and below the lesion. The tissue content of guanosine and its metabolic product guanine was measured by HPLC and CE analysis. Data are the mean ± SEM
New oligodendrocytes in injured spinal cords come from a population of endogenous progenitor cells present in the CNS that can differentiate into mature cells capable of myelinating axons [3, 12, 13, 56]. The data from the present study showed that the increase in the number of NG2+ cells is associated with an increase in Rip+ cells. Our data indicated that guanosine treatment induced an increase of mature oligodendroglia (Rip+ cells), likely due to proliferation and differentiation of their progenitor (NG2+) cells. Therefore, one would expect that Rip+ cells increase after NG2+ cells. However, this was not observed. But guanosine may well stimulate the proliferation of oligodendroglial progenitor cells at several stages of development. This would account for the observed results. Additionally, we cannot exclude that other possible mechanisms are involved in the process; for example, mature oligodendroglia could migrate from the adjacent normal tissue to the injury site soon after guanosine treatment.
Whether the effects on remyelination are due to guanosine, guanine or both is not clear. However, since a putative receptor for guanosine has been identified in the CNS, and since PNP (purine nucleoside phosphorylase) in CNS permits the interconversion of guanine and guanosine enabling guanine to act as a reservoir for guanosine, it is possible that guanosine may be responsible for causing the proliferation of the endogenous progenitor cells and the subsequent remyelination process, although this is by no means certain. But, since guanine itself is difficult to administer because of its poor solubility, administration of guanosine is the most effective delivery mechanism.
The mechanism by which remyelination is facilitated after chronic, traumatic SCI is unknown. Guanosine has been shown to stimulate the production and/or release of a variety of trophic factors [25]; for example bFGF, NGF and NT-3 have been demonstrated to increase the proliferation and survival of oligodendrocyte progenitors [3, 16, 57], induce oligodendroglial genesis [58–61], stimulate the proliferation of oligodendrocyte precursors and enhance remyelination after spinal cord traumatic injury [18, 19] and after toxin-induced demyelination [62]. There are no published reports of the effects of guanine in the vivo systems. However, in cultured cells, guanosine stimulates cell division apparently through a pertussis toxin-sensitive process involving MAP kinase [63]. Moreover, there is evidence of a G protein-coupled receptor in brain membranes that might be a candidate for a guanosine receptor [64]. Further studies will permit us to distinguish amongst these possibilities.
The present findings are the first to show that systemic treatment with guanosine after chronic SCI induces an improvement in function that is accompanied by the formation of mature oligodendrocytes. This effect of guanosine/guanine may be attributable to direct or indirect stimulation of endogenous oligodendrocyte lineage progenitors in the spinal cord and remyelination of axons at the injury site.
These findings raise the possibility that the administration of guanosine may be effective in treating spinal cord injuries and in the treatment of demyelinating diseases such as multiple sclerosis where quiescent progenitors exist in demyelinated plaques [65–70].
Abbreviations
- BBB:
-
Basso-Beattie-Bresnahan locomotor rating scale
- bFGF:
-
basic fibroblast growth factor
- BrdU:
-
bromodeoxyuridine
- CE:
-
capillary electrophoresis
- CNS:
-
central nervous system
- DAB:
-
diaminobenzidine
- FITC:
-
fluorescein isothiocyanate
- GFAP:
-
glial fibrillary acidic protein
- HPLC:
-
high-performance liquid chromatography
- MBP:
-
myelin basic protein
- NG2:
-
a chondroitin sulfate proteoglycan
- NGF:
-
nerve growth factor
- NT-3:
-
neurotrophin-3
- OFWT:
-
open field walking test
- PBS:
-
phosphate-buffered saline
- PFA:
-
paraformaldehyde
- PNP:
-
purine nucleoside phosphorylase
- PNS:
-
peripheral nervous system
- SCI:
-
spinal cord injury
- TdT:
-
terminal deoxynucleotidyltransferase
- TGF beta:
-
transforming growth factor beta
- TUNEL:
-
terminal deoxynucleotide transferase dUTP nick end labelling
References
Bunge MB, Bunge RP, Ris H (1961) Ultrastructural study of remyelination in an experimental lesion in adult cat spinal cord. J Biophys Biochem Cytol 10:67–94
Bunge RP, Puckett WR, Becerra JL, Marcillo A, Quencer RM (1993) Observations on the pathology of human spinal cord injury. A review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Adv Neurol 59:75–89
McDonald JW, Belegu V (2006) Demyelination and remyelination after spinal cord injury. J Neurotrauma 23:345–359
Carlson SL, Parrish ME, Springer JE, Doty K, Dossett L (1998) Acute inflammatory response in spinal cord following impact injury. Exp Neurol 151:77–88
Dusart I, Schwab ME (1994) A monoclonal antibody (IN-1) which neutralizes neurite growth inhibitory proteins in the rat CNS recognizes antigens localized in CNS myelin. J Neurocytol 23:209–217
Fleming CJ, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC (2006) The cellular inflammatory response in human spinal cords after injury. Brain 129:3249–3269
Hagg T, Oudega M (2006) Degenerative and spontaneous regenerative processes after spinal cord injury. J Neurotrauma 23:264–280
Blakemore WF, Franklin RJ (2000) Remyelinating the demyelinated CNS. Novartis Found Symp 231:289–298
Gilson J, Blakemore WF (1993) Failure of remyelination in areas of demyelination produced in the spinal cord of old rats. Neuropathol Appl Neurobiol 19:173–181
Ludwin SK (1988) Remyelination in the central nervous system and the peripheral nervous system. Adv Neurol 47:215–254
Smith EJ, Blakemore WF, McDonald WI (1979) Central remyelination restores secure conduction. Nature 280:395–396
Norton WT (1996) Do oligodendrocytes divide? Neurochem Res 21:495–503
Gensert JM, Goldman JE (1997) Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 19:197–203
Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, Gage FH (2000) Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci 20:2218–2228
McTigue DM, Wei P, Stokes BT (2001) Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci 21:3392–3400
Barres BA, Raff MC, Gaese F, Bartke I, Dechant G, Barde YA (1994) A crucial role for neurotrophin-3 in oligodendrocyte development. Nature 367:371–375
Rosano C, Felipe-Cuervo E, Wood PM (1999) Regenerative potential of adult O1+ oligodendrocytes. Glia 27:189–202
Stankoff B, Aigrot MS, Noel F, Wattilliaux A, Zalc B, Lubetzki C (2002) Ciliary neuro-trophic factor (CNTF) enhances myelin formation: a novel role for CNTF and CNTF-related molecules. J Neurosci 22:9221–9227
McTigue DM, Horner PJ, Stokes BT, Gage FH (1998) Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci 18:5354–5365
Baumann N, Pham-Dinh D (2001) Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 81:871–927
David S, Lacroix S (2003) Molecular approaches to spinal cord repair. Annu Rev Neurosci 26:411–440
Horner PJ, Gage FH (2000) Regenerating the damaged central nervous system. Nature 407:963–970
Yan H, Wood PM (2000) NT-3 weakly stimulates proliferation of adult rat O1(−)O4(+) oligodendrocyte-lineage cells and increases oligodendrocyte myelination in vitro. J Neurosci Res 62:329–335
Goddard DR, Berry M, Kirvell SL, Butt AM (2001) Fibroblast growth factor-2 inhibits myelin production by oligodendrocytes in vivo. Mol Cell Neurosci 18:557–569
Rathbone MP, Middlemiss PJ, Gysbers JW, Andrew C, Herman MA, Reed JK, Ciccarelli R, Di Iorio P, Caciagli F (1999) Trophic effects of purines in neurons and glial cells. Prog Neurobiol 59:663–690
Gysbers JW, Guarnieri S, Mariggio MA, Pietrangelo T, Fano G, Rathbone MP (2000) Extracellular guanosine 5′ triphosphate enhances nerve growth factor-induced neurite outgrowth via increases in intracellular calcium. Neuroscience 96:817–824
Di Iorio P, Caciagli F, Giuliani P, Ballerini P, Ciccarelli R, Sperling O, Zoref-Shani E, Benowitz L, Traversa U, Bombi G, Fiorio T, D’Alimonte I, Andrew CM, Crocker CE, Werstiuk ES, Middlemiss PJ, Rathbone MP (2001) Purine nucleosides protect injured neurons and stimulate neuronal regeneration by intracellular and membrane receptor-mediated mechanisms. Drug Dev Res 52:303–315
Ciccarelli R, Di Iorio P, D’Alimonte I, Giulani P, Florio T, Caciagli F, Middlemiss PJ, Rathbone MP (2000) Cultured astrocyte proliferation induced by extracellular guanosine involves endogenous adenosine and is raised by the co-presence of microglia. Glia 29:202–211
Gysbers JW, Rathbone MP (1992) Guanosine enhances NGF-stimulated neurite outgrowth in PC12 cells. Neuroreport 3:997–1000
Gysbers JW, Rathbone MP (1996) GTP and guanosine synergistically enhance NGF-induced neurite outgrowth from PC12 cells. Int J Dev Neurosci 14:19–34
Jiang S, Khan MI, Lu Y, Wang J, Buttigieg J, Werstiuk ES, Ciccarelli R, Caciagli F, Rathbone MP (2003a) Guanosine promotes myelination and functional recovery in chronic spinal injury. Neuroreport 14:2463–2467
Blight AR (1991) Morphometric analysis of a model of spinal cord injury in guinea pigs, with behavioral evidence of delayed secondary pathology. J Neurol Sci 103:156–171
Gruner JA, Yee AK, Blight AR (1996) Histological and functional evaluation of experimental spinal cord injury: evidence of a stepwise response to graded compression. Brain Res 729:90–101
Jiang S, Wang J, Khan MI, Middlemiss PJ, Salgado-Ceballos H, Werstiuk ES, Wickson R, Rathbone MP (2003b) Enteric glia promote regeneration of transected dorsal root axons into spinal cord of adult rats. Exp Neurol 181:79–83
Jiang S, Khan MI, Middlemiss PJ, Lu Y, Werstiuk ES, Crocker CE, Ciccarelli R, Caciagli F, Rathbone MP (2004) AIT-082 and methylprednisolone singly, but not in combination, enhance functional and histological improvement after acute spinal cord injury in rats. Int J Immunopathol Pharmacol 17:353–366
Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12:1–21
Basso DM, Beattie MS, Bresnahan JC (1996) Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol 139:244–256
Bruck W, Kuhlmann T, Stadelmann C (2003) Remyelination in multiple sclerosis. J Neurol Sci 206:181–185
Friedman B, Hockfield S, Black JA, Woodruff KA, Waxman SG (1989) In situ demonstration of mature oligodendrocytes and their processes: an immunocytochemical study with a new monoclonal antibody, rip. Glia 2:380–390
Ciccarelli R, Di Iorio P, Giuliani P, D’Alimonte I, Ballerini P, Caciagli F, Rathbone MP (1999) Rat cultured astrocytes release guanine-based purines in basal conditions and after hypoxia/hypoglycemia. Glia 25:93–98
Keirstead HS, Blakemore WF (1997) Identification of post-mitotic oligodendrocytes incapable of remyelination within the demyelinated adult spinal cord. J Neuropathol Exp Neurol 56:1191–1201
Redwine JM, Armstrong RC (1998) In vivo proliferation of oligodendrocyte progenitors expressing PDGFalphaR during early remyelination. J Neurobiol 37:413–428
Polito A, Reynolds R (2005) NG2-expressing cells as oligodendrocyte progenitors in the normal and demyelinated adult central nervous system. J Anat 207:707–716
Dawson MR, Polito A, Levine JM, Reynolds R (2003) NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci 24:476–488
Carroll WM, Jennings AR, Ironside LJ (1998) Identification of the adult resting progenitor cell by autoradiographic tracking of oligodendrocyte precursors in experimental CNS demyelination. Brain 121:293–302
Keirstead HS, Levine JM, Blakemore WF (1998) Response of the oligodendrocyte progenitor cell population (defined by NG2 labelling) to demyelination of the adult spinal cord. Glia 22:161–170
Levine JM, Reynolds R (1999) Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Exp Neurol 160:333–347
Lytle JM, Wrathall JR (2007) Glial cell loss, proliferation and replacement in the contused murine spinal cord. Eur J Neurosci 25:1711–1724
Williams A, Piaton G, Lubetzki C (2007) Astrocytes—friends or foes in multiple sclerosis? Glia 55:1300–1312
Di Iorio P, Kleywegt S, Ciccarelli R, Traversa U, Andrew CM, Crocker CE, Werstiuk ES, Rathbone MP (2002) Mechanisms of apoptosis induced by purine nucleosides in astrocytes. Glia 38:179–190
Di Iorio P, Ballerini P, Traversa U, Nicoletti F, D’Alimonte I, Kleywegt S, Werstiuk ES, Rathbone MP, Caciagli F, Cicarelli R (2004) The antiapoptotic effect of guanosine is mediated via the activation of PI3-kinase/Akt/PKB pathway in cultured rat astrocytes. Glia 46:356–368
Pettifer KM, Kleywegt S, Bau CJ, Ramsbottom JD, Vertes E, Ciccarelli R, Caciagli F, Werstiuk ES, Rathbone MP (2004) Guanosine protects SH-SY5Y cells against beta-amyloid-induced apoptosis. Neuroreport 15:833–836
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8:963–970
Zhang R, Xue YY, Lu SD, Wang Y, Zhang LM, Huang YL, Signore AP, Chen J, Sun FY (2006) Bcl-2 enhances neurogenesis and inhibits apoptosis of newborn neurons in adult rat brain following a transient middle cerebral artery occlusion. Neurobiol Dis 24:345–356
Mason JL, Jones JJ, Taniike M, Morell P, Suzuki K, Matsushima GK (2000) Mature oligodendrocyte apoptosis precedes IGF-1 production and oligodendrocyte progenitor accumulation and differentiation during demyelination/remyelination. J Neurosci Res 61:251–262
Vick RS, Neuberger TJ, Devries GH (1992) Role of adult oligodendrocytes in remyelination after neural injury. J Neurotrauma 9:S93–S103
Cohen RI, Marmur R, Norton WT, Mehler MF, Kessler JA (1996) Nerve growth factor and neurotrophin-3 differentially regulate the proliferation and survival of developing rat brain oligodendrocytes. J Neurosci 16:6433–6442
Fressinaud C, Vallat JM, Labourdette G (1995) Basic fibroblast growth factor down-regulates myelin basic protein gene expression and alters myelin compaction of mature oligodendrocytes in vitro. J Neurosci Res 40:285–293
McMorris FA, Mozell RL, Carson MJ, Shinar Y, Meyer RD, Marchetti N (1993) Regulation of oligodendrocyte development and central nervous system myelination by insulin-like growth factors. Ann N Y Acad Sci 692:321–334
McMorris FA, McKinnon RD (1996) Regulation of oligodendrocyte development and CNS myelination by growth factors: prospects for therapy of demyelinating disease. Brain Pathol 6:313–329
Bansal R, Pfeiffer SE (1997) FGF-2 converts mature oligodendrocytes to a novel phenotype. J Neurosci Res 50:215–228
Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP (2001) TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci 4:1116–1122
Rathbone MP, Pilutti L, Caciagli F, Jiang S (in press) Neurotrophic effects of extracellular guanosine (review). Nucleosides Nucleotides Nucleic Acids
Traversa U, Bombi G, Di Iorio P, Ciccarelli R, Werstiuk ES, Rathbone MP (2002) Specific [(3)H]-guanosine binding sites in rat brain membranes. Br J Pharmacol 135:969–976
Wolswijk G (1998) Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci 18:601–609
Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD (2000) NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 20:6404–6412
Chang A (2002) Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 346:165–173
Scolding N, Franklin R, Stevens S, Heldin CH, Compston A, Newcombe J (1998) Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain 121:2221–2228
Wilson HC, Scolding NJ, Raine CS (2006) Co-expression of PDGF alpha receptor and NG2 by oligodendrocyte precursors in human CNS and multiple sclerosis lesions. J Neuroimmunol 176:162–173
Nait-Oumesmar B, Picard-Riera N, Kerninon C, Decker L, Seilhean D, Hoglinger GU, Hirsch EC, Reynolds R, Baron-Van Evercooren A (2007) Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc Natl Acad Sci U S A 104:4694–4699
Acknowledgements
We thank Dr. Raelene Kinlough-Rathbone for her helpful suggestions and critical appraisal of the manuscript, and Drs. Richard Borgens, Andrew Blight, and John Gruner for advice on the spinal crush model. We are grateful to Prof. Marie T. Filbin, Department of Biological Sciences, CUNY, NY, for kindly providing the anti-P0.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Jiang, S., Ballerini, P., Buccella, S. et al. Remyelination after chronic spinal cord injury is associated with proliferation of endogenous adult progenitor cells after systemic administration of guanosine. Purinergic Signalling 4, 61–71 (2008). https://doi.org/10.1007/s11302-007-9093-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-007-9093-8