
Abstract
Small molecule permeability through cellular membranes is critical to a better understanding of pharmacodynamics and the drug discovery endeavor. Such permeability may be estimated as a function of the free energy change of barrier crossing by invoking the barrier domain model, which posits that permeation is limited by passage through a single “barrier domain” and assumes diffusivity differences among compounds of similar structure are negligible. Inspired by the work of Rezai and co-workers (JACS 128:14073–14080, 2006), we estimate this free energy change as the difference in implicit solvation free energies in chloroform and water, but extend their model to include solute conformational affects. Using a set of eleven structurally diverse FDA approved compounds and a set of thirteen congeneric molecules, we show that the solvation free energies are dominated by the global minima, which allows solute conformational distributions to be effectively neglected. For the set of tested compounds, the best correlation with experiment is obtained when the implicit chloroform global minimum is used to evaluate the solvation free energy difference.
Similar content being viewed by others


Introduction
The ability to cross a lipid bilayer is an essential character that all drugs must posses. For example, a compound that readily diffuses through the intestinal enterocyte boundary is far more likely to be administered orally than a compound that does not. Topically administered drugs, a mainstay in dermatology, must often pass through several epidermal layers and their associated membranes [1]. Likewise, pulmonary administered compounds, such as those used in the treatment of Cystic Fibrosis [2], must cross the alveolar epithelium and capillary endothelium membranes before entering systematic circulation. Generally, with the exception of those targeting blood-borne elements, all drugs must cross capillary membranes before exiting systematic circulation and reaching their cellular target, regardless of the administration route [3]. Even drugs targeting blood platelets must pass through a cellular membrane [3].
Experimental methods of measuring in vitro small molecule membrane permeability commonly include the cell based Caco-2 assay [4] and the artificial membrane based PAMPA [5] assays. Caco-2 assays utilize a cell line derived from human colon carcinoma, have characteristics that resemble intestinal epithelial cells, including intercellular junctions and transport proteins, and can yield permeabilities with good correlation to human oral drug absorption [6]. Parallel Artificial Membrane Permeability Assays (PAMPA), on the other hand, are a lower-cost, higher-throughput alternative that utilize an artificial membrane generally composed of a mixture of lipids dissolved in an organic solvent, embedded in a porous filter. As PAMPA lack intercellular junctions and active transporters, permeability is exclusively passive. Despite the difference in cell and artificial membrane based assays, correlation between the two can be good [7].
Computational methods of predicting small molecule membrane permeability are fundamentally important in drug lead generation and optimization and are an actively evolving area of methodological development. For example, compound permeability estimates allow one to select a compound library subset for docking, or to determine an appropriate lead optimization strategy. While experimental permeability measurements are ideal, they are often too slow or expensive to filter the ~104–106 compounds typical of a docking screen, and they require that a lead series be synthesized before assay. Alternatively, computational models can be rapid, often correlate well with experiment, and range from the detailed and expensive molecular dynamics (MD) methods [8, 9], to the less accurate but inexpensive knowledge-based quantitative structural permeability relationship (QSPR) methods [10].
In 2006, the Jacobson group published results from a unique physics-based method, intermediate in detail between the MD and QSPR methods [11] that is loosely based on the homogenous solubility model of passive membrane permeability, \( P = (D/d)K_{p} \). The permeability is P, (cm/s), the diffusion coefficient is D, (cm2/s), the membrane thickness is d, (cm), and K p is the unitless partition coefficient of the drug between bulk water and the membrane, which is taken as a homogenous low-dielectric slab. By invoking the barrier domain model [12], which posits that permeation is limited by passage through a single “barrier domain,” and assuming diffusivity differences are small, the model determines permeability as a function of the free energy change of barrier crossing. The free energy change of barrier crossing is estimated by the difference in implicit solvent chloroform and water solvation free energies, evaluated using a single member of the conformational ensemble found at the global minimum in the chloroform solvent. The choice of implicit solvent, while not necessarily the most accurate, captures the fundamental dielectric character of the membrane and water phases but incurs significantly less computational cost than an all-atom method. Consequently, the use of implicit solvent offers a unique balance between accurate, high-cost, low throughput all-atom methods, and less accurate, lower-cost higher throughput methods. While the model worked well for a set of cyclic peptides [11], performance deteriorated for two small sets of drugs [11, 13]. This led us to explore whether a more rigorous treatment of conformational distributions would improve accuracy and how sensitive the method is to subtle structural differences characteristic of lead optimization.
In this work, we investigate the affects of using a more rigorous conformational distribution treatment by comparing three different configuration integral approximations. We also expanded their study to include a set of simple benzene congeners to determine how sensitive the model is to subtle changes that might be proposed during lead optimization. Moreover, we compare the performance of each approximation to those of QikProp, a fast, easy to use, knowledge-based pharmacokinetic property predictor available from Schrödinger [14]. In all cases, predictions are compared to PAMPA data.
Methods and theory
Relating the homogenous solubility and barrier domain models of membrane permeability
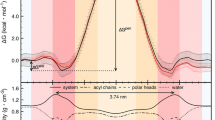
The barrier domain model of membrane permeability rationalizes treatment of structurally anisotropic membranes as isotropic homogenous slabs. The barrier domain model, which is consistent with both experiment [12] and computation [8, 9], posits that for polar compounds, permeability is limited by passage through the highly ordered membrane region immediately behind the acyl-chain head group linkages. In this region, solubility is low due to the apolar character of the acyl-hydrocarbon chain, and diffusivity is low due to the ordered structure of the chains. Both characteristics give rise to high permeability resistance, making passage through this region rate limiting. The free energy change associated with barrier crossing is then given by the difference in free energy of the ensemble of conformations accessible at the hydrated head groups, which constitute the membrane-water interface, and those accessible at the transition state in the ordered acyl-chain membrane region. In this way, the homogenous solubility model of passive membrane permeability is similar to transition state theory, i.e. \( P = (D/d)e^{{ - \Updelta {\text{G}}/RT}} \) where ΔG is the free energy of barrier crossing, and the diffusivity pre-factor may be assumed to roughly cancel across sets of similar compounds. In this work, as in the work reported by the Jacobson group, the free energy barrier is estimated as the difference in chloroform and aqueous solvation free energies. In the next section, we develop a statistical thermodynamic expression for this free energy change, which leads to three approximations.
Statistical thermodynamic formulation
If differences in intra-membrane diffusivity are neglected, according to the homogenous solubility diffusion and barrier domain models, the logged permeability of one molecule relative to another is,
where β = (RT) −1, with R the universal gas constant and T the temperature. The calculated quantity, ∆G, is the difference in solvation free energies of a molecule dissolved in water and in the membrane. Using an implicit chloroform solvent to model the membrane, implicit water to model an aqueous solution, and assuming the difference in volume between the chloroform and aqueous solutions is negligibly small, ∆G is given,
U( x ) is the potential energy of the ligand in a conformation given by its coordinates x. \( W_{{{\text{H}}_{ 2} {\text{O}}}} ({\mathbf{x}}) \) is the aqueous solvation free energy, or solvation potential of mean force [15], when the ligand is held in a conformation x; a similar definition holds for \( W_{{{\text{CHCl}}_{ 3} }} ({\mathbf{x}}) \). The solvation free energies are further decomposed into electrostatic contributions that arise from the difference in free energies of charging the ligand in the solvent and gas phases, respectively, and a non-polar contribution that results from creating a solute sized cavity in the solvent [16].
While the configuration integrals in Eq. 2 are evaluated over all possible ligand conformations, the Boltzmann factors in each are largest for conformations with low potential and solvation free energies. Consequently, by increasing the extent of conformational sampling around the global minimum, each integral can be approximated with increasing accuracy. Three approximations are discussed. The first, which we denote “the single-state approximation,” is consistent with the calculations reported by the Jacobson group [11, 13]. In the single-state approximation, it’s assumed that the global minimum conformation in chloroform dominates both integrals. This assumption implies that the conformation that resides at the global minimum in chloroform is the same conformation that resides at the global minimum in water. If the global minimum in chloroform is designated, \( {\mathbf{x}}_{{{\text{CHCl}}_{ 3} }} \), then applying the single-state approximation to Eq. 2 yields,
In the second, which we denote “the two-state approximation,” it is assumed that the global minimum in each solvent dominates their respective integrals. If the global minimum in water is designated \( {\mathbf{x}}_{{{\text{H}}_{ 2} {\text{O}}}} \), and the global minimum in chloroform is designated as above, then applying the two-state approximation to Eq. 2 gives,
Finally, in the third approximation, which we denote “the predominate-states approximation,” we assume that conformations in addition to the global minima contribute significantly to each integral. Predominate states methods are well known, and have been used successfully to study the thermodynamics of ligand binding in host–guest systems [17] and proteins [18]. Characteristically, the methods decompose the configuration integral into a sum of local configuration integrals, each centered on a minimum energy conformation enumerated during a conformation search. The configuration integrals are evaluated by normal mode analysis in either bond-angle-torsion (BAT) coordinates [19] or in the rigid-rotor-harmonic oscillator coordinate system (RRHO) [20, 21]. In this work, the RRHO system is used. The RRHO separates the “internal” normal coordinate vibrations from the “external” translations and rotations, which leads to rotational momentum free energy contributions and, along with the predominate states approximation, allows an approximation to Eq. 2 to be written,
I a,i I b,i I c,i are the principal moments of inertia of the ith conformation, similar definitions hold for I a,j I b,j I c,j . The r coordinates are the 3L-6 internal coordinates of the ligand, where L is the number of ligand atoms. Each integral is evaluated using Monte Carlo importance sampling, following the “mode integration,” or MINTA approach [20]. Taking the chloroform integrals, for example,
where the terms enclosed in 〈〉 denote an average over the distribution p i (r) contained in the volume element V i . The average is numerically approximated by randomly sampling M solute conformations from the multivariate Gaussian produced by normal mode analysis in the RRHO coordinate system,
Similar definitions and procedures exist for the integrals evaluated in water.
Small molecule test set
We performed our calculations on two sets of small molecules. The first, which we denote the “FDA set,” is a structurally diverse set of eleven weakly basic FDA approved compounds whose “intrinsic” permeabilities, i.e. permeabilities if all molecules were in their uncharged form, were estimated by fitting PAMPA data using the \( {\text{pK}}_{a}^{\text{flux}} \) method [7]. These eleven compounds are the same as those used by the Jacobson group and allow a direct comparison of our results [13]. These compound structures are shown in Fig. 1.

Structures and names of the 11 compounds in the FDA set
To test the sensitivity of the calculations to the subtle structural changes typical of lead optimization, we selected a congeneric series of thirteen phenol and aniline derivatives, which we denote the “congeneric set,” whose structures are shown in Fig. 2. The “effective” permeabilities of these compounds were measured by PAMPA [22]. Unlike intrinsic permeability, effective permeability does not correct for the presence of charged species if the compound is acidic or basic. Assuming that only the uncharged species can cross the lipid bilayer, effective permeabilities will always be less than intrinsic permeabilities for titratable compounds. This can be problematic in some instances. For example, two compounds with very different pKa values can have very different effective permeabilities, despite having identical intrinsic permeabilities. The aniline and phenol derivatives in the congeneric set have roughly the same proportion of uncharged species in solution, however, the discrepancy between effective and intrinsic permeabilities is not expected to have gross, erroneous effects on the relative permeability calculations that we describe. For both compound sets, all of the calculations were performed with the compounds in their neutral, or uncharged, states.

Structures and names of the 13 compounds in the congeneric set
Computational methods
Monte Carlo (MC) conformational searches were carried out using the MC Multiple Minimum program in v9.7 of MacroModel within the Schrödinger molecular modeling suite [23]. Initial coordinates for the FDA set were obtained from PDB files found at the drug databank (drugbank.ca). Initial coordinates for the congeneric set were constructed using the modeling facilities in maestro [24]. A 10,000 step MC conformational search was performed using a generalized Born surface area (GBSA) solvent model, taking parameters that match either water or chloroform. As in the previously described works [11, 13], solute force field parameters were described using the 2005 OPLS parameter set, which has been enhanced by Schrödinger to provide a larger coverage of organic functionality. MC was performed by randomly varying between 1 and 5 torsions, over a range of angles between 0 and 180°, producing a trial conformation. Trial conformations were minimized for 1,000 steps using Polak-Ribiere conjugate gradient minimization. Minimized trial structures were retained if their energy was less than 12 kcal mol−1 above the current global minimum, and if the distance separating any one heavy atom (or hydroxyl or thiol hydrogen atom) on the minimized trial conformation was greater than 0.5 Å from the position of the equivalent heavy atom (or hydroxyl or thiol hydrogen atom) on each of the previously retained conformations after a rigid-body least squares fitting. MC trial conformations were initiated from the least-used structure, a strategy that has previously been shown to increase the convergence rate.
Successfully determining the global minimum in a given solvent requires exhaustively searching the conformational landscape, an effort that can be frustrated if large energetic barriers separate adjacent regions. To ensure adequate conformational sampling, seven additional MC searches were performed for each solute in each solvent, and the global minima were extracted from the combined search results after removing redundant conformations. The first four additional searches were initiated from conformations at energy intervals of 0 to 1, 1 to 2, 2 to 3 and 3 to 5 kcal mol−1 above the global minimum determined in the first search. The remaining 3 searches were initiated from conformations with energies greater than 5 kcal mol−1 above the global minimum of the first search. When possible, each conformation was separated by a least 1 kcal mol−1 from the others. This is similar to multi-copy molecular dynamics simulations, which have been shown to increase conformational sampling in the protein crambin [25] and produce greater statistical precision in simulations of the RN24 peptide [26]. In a conformational search, adequate convergence occurs when additional searching does not contribute new, unique conformations. Convergence may be monitored by plotting the total number of conformations as a function of the number of MC searches performed, which was done for the compounds in the FDA set (supplementary Fig. 1); as fewer new conformations are found, these plots should asymptotically approach a fixed value, indicating convergence. As compounds in the congeneric set have fewer rotatable bonds, we only performed a single conformational search, as outlined above, and assumed that it was sufficient for convergence. In all conformational searches, symmetrically equivalent conformations were retained and no electrostatic cutoffs were used.
Following the MC searches, each conformation was re-minimized using 1,500 steps of truncated Newton conjugate gradient minimization to determine the global minimum. For the single-state approximation, the terms in Eq. 3 were evaluated by calculating the solvation PMF with GBSA models of water and chloroform using the global minimum conformation determined in chloroform.
The change in free energy for the two-state approximation was evaluated similarly, but the global minimum determined in water was also used and the free energy difference was evaluated according to Eq. 4. For both the single and two-state approximations, energy evaluations were carried out using the “current energy” function in MacroModel, which is accessible through Schrödinger’s Maestro interface or the command line. In all energy evaluations, no electrostatic cutoffs were used.
MINTA [20, 23] was used to evaluate each unique conformation after the results of all eight MC conformational searches were combined. Importance sampling integration was carried out according to Eq. 7 for the ten normal coordinates with the lowest vibrational frequencies, while the analytical expression of the configuration integral over the remaining modes was determined. The integration volume, V i , for the numerically integrated modes was chosen as the smaller of ±3σ or ±3 Å, in order to reduce large inaccuracies that may arise if the potential energy along one or more of the numerically integrated modes exhibits extensive anharmonicity [20]. Additionally, the choice decreases the likelihood that the integration volumes of adjacent states will overlap, resulting in double counting [20]. As in the MC conformational search, a combination of the Schrödinger-extended 2005 OPLS force field and GBSA implicit solvent were used to describe the energy surface. The principal moments of inertia for each minimum-energy conformation were determined in a post-processing step within Matlab [27] by diagonalizing the associated unit-mass inertial tensor. Unit masses were used to be consistent with the unit-mass Hessian, which MINTA employs when determining the sampling distributions [27].
QikProp v3.2 was used to predict Caco-2 permeabilities [14]. As QikProp does not predict PAMPA permeabilities, only Caco-2 permeabilities are reported. QikProp permeability calculations are conformationally invariant, and for both the FDA and congeneric set, starting conformations were used without minimization.
Results and discussion
The FDA set
The logarithm of the experimentally determined permeabilities are plotted against the corresponding ∆G values determined using the single-state, two-state, and predominate-states approximations in Fig. 3a, b and c, respectively. The QSPR results are shown in Fig. 3d. The single-state approximation has an R2 of 0.75, the two-state approximation an R2 of 0.68, the predominant-states approximation an R2 of 0.71, and the QSPR method predictions have an R2 of 0.75. The single-state approximation results are very slightly better than the values previously reported by Kalyanaraman and Jacobson [13], who carried out a single conformational search, which may not have converged to the true global minimum. For example, plotting the linear-correlation coefficient as a function of the number of independent MC searches combined to find the global minimum conformation (supplementary Fig. 2) shows that after the 5th conformational search, a new global minimum is found, and the linear correlation improves.

Correlation with experiment for compounds in the FDA set, a the single-state approximation R2 = 0.75, b the two-state approximation R2 = 0.68, c the predominant-states approximation R2 = 0.71, d QSPR R2 = 0.75
The relative contributions to the free energy of those conformations near the global minimum bears on the calculated outcomes. To estimate this quantity, we calculated the fraction of free energy due to conformations within 1.00 kcal mol−1 of the global minimum, as well as the factional contribution of just the global minimum. Reported in Table 1, these values indicate that despite the hundreds to thousands of conformations in the ensemble (supplementary Fig. 1), the global minima constitute 97 to 99% of the total free energy, while conformations within 1.00 kcal mol−1 comprise greater than 99% of the total. This shows that sampling additional contributions beyond the global minima does little to alter the value of the calculated solvation free energies, partly explaining why the predominant-states approximation did not significantly improve upon the single- and two-state approximations.
As an additional consideration, we analyzed the structural differences of the global minimum conformations in membrane and in water and estimated \( \Updelta {\text{G}}_{{{\text{H}}_{ 2} {\text{O}}}} \) [13], defined as the difference in free energy between the global minimum conformation in water, \( {\mathbf{x}}_{{{\text{H}}_{ 2} {\text{O}}}} \), and the global minimum conformation in chloroform, \( {\mathbf{x}}_{{{\text{CHCl}}_{3} }} \), each evaluated in water, which measures the reorganizational free energy upon solvent transfer, i.e.,
Global minimum conformations for representative compounds are shown in Fig. 4 while the heavy-atom and polar-hydrogen atom RMSD separating the global minima, along with the \( \Updelta {\text{G}}_{{{\text{H}}_{ 2} {\text{O}}}} \) values are reported in Table 1. Most of the RMSD values are under 2 Å, indicating a high degree of similarity in chloroform and water, which agrees with the images presented in Fig. 4. This is consistent with the \( \Updelta {\text{G}}_{{{\text{H}}_{ 2} {\text{O}}}} \) values that are all nearly less than RT at 310 K, indicating that in water, the conformation at the global minimum in chloroform is frequently visited. The notable exception is verapamil, which is also the largest outlier in the single-state approximation (Fig. 3a). Based on the barrier domain model, the slightly better performance of the single-state model implies that the conformations found at the global minimum in implicit chloroform may better represent the dominant conformations found at the membrane-water interface. In contrast, the fact that verapamil is the largest outlier and has the largest \( \Updelta {\text{G}}_{{{\text{H}}_{ 2} {\text{O}}}} \) value may indicate that its dominant membrane-water interface conformation is more similar to the global minimum in bulk water.

Global minima in chloroform and water for representative compounds from the FDA set, a alfentanil, b verapamil
The congeneric set
The logarithm of the experimentally determined permeabilities are plotted against the corresponding ∆G values determined using the single-, two- and predominate-states approximations (Fig. 5a, b and c, respectively). The QSPR results are shown in Fig. 4d. The single-state approximation gives an R2 of 0.72, the two-state approximation an R2 of 0.72, the predominant-states approximation an R2 of 0.71, and the QSPR method predictions have an R2 of 0.58.

Correlation with experiment for compounds in the congeneric set, a the single state approximation R2 = 0.72, b the two-state approximation, R2 = 0.72, c the predominant-states approximation R2 = 0.71, d QSPR R2 = 0.58
The single- and two-state approximations performed identically. The matching performance is attributable to the small number of rotamers in the congeneric set, which results in a very small conformational ensemble and virtually identical global minimum conformations in each solvent. ΔGwat and heavy atom RMSD values (Table 2) support this claim. The absolute value of ΔGwat doesn’t exceed 0.16 kcal mol−1 and all of the RMSD values are below 0.38 Å, with most falling below 0.01 Å, confirming the near conformational parity of the global minima in each solvent.
The small conformational ensemble also explains the performance of the predominant-states approximation, which was nearly equivalent to the single-state and two-state approximations. When there are a substantial number of conformations that contribute to the configuration integral, the free energies estimated using the predominant-states approximation will be different than those estimated using only the global minimum. However, as compounds in the congeneric set have few rotamers, the number of conformations is limited. For example, we found two phenol conformations in both chloroform and water, while for BPMC, the most flexible of the set, 41 conformations were found in chloroform and 44 in water. This is significantly fewer than the hundreds to thousands of conformers found for compounds in the FDA set. As a result, the three approximations yield similar estimates, explaining why all three give permeabilities that correlate nearly identically with experiment.
None of the approximations show particular sensitivity to the small structural differences present in the congeneric set. Rather, with the exception of the two-state approximation, performance is slightly worse on this set than the structurally more diverse FDA set. Nevertheless, all three approximations outperform the QSPR model, which seems unable to effectively separate the permeabilities of the structural homologs (note the mass of data points on the upper right hand side of the curve in Fig 5d). Assuming the relative performance extends beyond the simple series examined here, each of the approximations may be better at teasing apart the subtle structural influences on permeability than an all-purpose QSPR method. In particular, when the number of rotamers is small, the approximations perform nearly identically, and as the single-state approximation is the least computationally intensive, it is the most appropriate choice.
Conclusions
This work expanded upon earlier work by the Jacobson group [11, 13] and addressed the effects of conformational sampling on permeability predictions based on the homogenous solubility model of passive membrane permeability. By invoking the barrier domain model [12], which posits that permeation is limited by passage through a single “barrier domain” and assumes diffusivity differences are small, the model determines permeability as a function of the free energy change of barrier crossing. The barrier domain is generally taken as the ordered, apolar region behind the head groups [8, 9, 12], and in this work, as in the work reported by the Jacobson group, the free energy barrier is estimated as the difference in chloroform and aqueous solvation free energies. A statistical thermodynamic description of the solvation free energy difference was developed and three limiting assumptions, differing in the extent of solute conformational sampling, were made to evaluate the configuration integrals. The single-state approximation, which is identical to the approximation used by the Jacobson group [11, 13], is based on the assumption that the global minimum conformation in chloroform dominates both integrals. The two-state approximation is based on the assumption that the global minimum in each solvent dominates their respective integrals. Finally, the predominant-states approximation decomposes each configuration integral into a sum of configuration integrals, each of which are centered on conformers determined in a conformational search, that are evaluated by Monte Carlo importance sampling. We tested each approximation on a set of eleven structurally diverse FDA approved compounds, as well as a set of thirteen simple benzene congeners intended to mimic the subtle changes that might be proposed during lead optimization. Moreover, to compare the performance of each approximation to a QSPR model, permeabilities for both sets were computed with QikProp, a fast, knowledge-based pharmacokinetic property predictor available from Schrödinger.
Our principal finding was that more rigorous treatments of the conformational distributions of the solute do not improve correlation with experiment. We attribute this to the solvation free energy landscape, which for all compounds in both chloroform and water is dominated by the global minimum. Moreover, as the number of rotameric states decreases, the size of the conformational ensemble is reduced and discrepancy between the three approximations is diminished. Whether or not other small molecules behave similarly will need to be addressed on a case-by-case basis. Between the two-state and single-state approximation, the single-state approximation is likely the most robust, slightly outperforming the other two approximations on the FDA set, and performing as well as the two-state approximation on the congeneric set. While the QSPR model performed as well as the single-state model for the structurally diverse FDA set, it was unable to effectively separate the permeabilities of the congeneric set. Assuming the performances extend beyond the compound sets we’ve tested here, the computationally inexpensive QSPR methods are best applied as filters for large sets of structurally diverse compounds when establishing a docking library, while the more expensive single-state approximation may be suited to a lead-optimization setting.
Considering both the FDA and congeneric sets, coefficients of determination (R2) values were never better than 0.75, so while the estimated solvation free energies capture most of the permeability variation, some is lost, which may be attributable to one or more causes. First, it may indicate that the barrier domain model is an inappropriate description of membrane permeability. Structural anisotropy in the membrane is widely accepted, and it has been shown that both the free energy of solvating a compound in the membrane, as well as a compound’s intramembrane diffusion coefficient, depend on the depth of the compound in the membrane [8, 9]. As permeability is, in fact, due to passage across the entire membrane, and not just the rate limiting region, or “barrier-domain,” the barrier domain model falls short of a complete physical description of the process, possibly leading to erroneous estimates. Alternatively, the barrier domain model may adequately describe the fundamental quantities that affect membrane permeability, but errors in our calculations are causing the discrepancies with experiment. As generalized Born solvent models estimate results of the theoretically more rigorous Poisson-Boltzmann equation [28], which itself is a mean field approximation of the exact case, some of the calculations presented here may very well be erroneous. Unfortunately, as chloroform-water partition coefficients for the tested compounds have not been published, accurate calculations cannot currently be separated from inaccurate calculations. Nevertheless, the rapid GBSA implicit solvent models of water and chloroform require less computational effort than the more detailed all-atom models while still capturing the general dielectric character of the aqueous and membrane phases. A third alternative is that the compounds examined diffuse very differently through the membrane, invalidating our assumption that diffusion can be neglected. While any of these possibilities may explain why better correlation with experiment was not obtained, all three likely contribute to a greater or lesser extent depending upon the compound set of interest.
Finally, while the models we tested here can provide reasonable correlation with experiment, their robustness needs to be explored across much larger compound sets. However, as many drug-like molecules are acidic or basic, a robust means of treating titratable compounds first needs to be established.
References
Jensen JM, Proksch E (2009) The skin’s barrier. G Ital Dermatol Venereol 144(6):689–700
Courrier HM, Butz N, Vandamme TF (2002) Pulmonary drug delivery systems: recent developments and prospects. Crit Rev Ther Drug Carrier Syst 19(4–5):425–498
Minneman KP, Wecker L, Larner J, Brody TM (2005) Brody’s Human Pharmacology: Molecular to Clinical 4th edn. Elsevier Mosby, Philadelphia, PA
Cheng KC, Li C, Uss AS (2008) Prediction of oral drug absorption in humans—from cultured cell lines and experimental animals. Expert Opin Drug Metab Toxicol 4(5):581–590. doi:10.1517/17425255.4.5.581
Avdeef A (2005) The rise of PAMPA. Expert Opin Drug Metab Toxicol 1(2):325–342. doi:10.1517/17425255.1.2.325
Artursson P, Karlsson J (1991) Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun 175(3):880–885
Avdeef A, Artursson P, Neuhoff S, Lazorova L, Grasjo J, Tavelin S (2005) Caco-2 permeability of weakly basic drugs predicted with the double-sink PAMPA pKa(flux) method. Eur J Pharm Sci 24(4):333–349. doi:10.1016/j.ejps.2004.11.011
Marrink SJ, Berendsen HJC (1994) Simulation of water transport through a lipid membrane. J Phys Chem 98:4155–4168
Marrink SJ, Berendsen HJC (1996) Permeation process of small molecules across lipid membranes studied by molecular dynamics simulations. J Phys Chem 100(41):16729–16738
Akamatsu M, Fujikawa M, Nakao K, Shimizu R (2009) In silico prediction of human oral absorption based on QSAR analyses of PAMPA permeability. Chem Biodivers 6(11):1845–1866. doi:10.1002/cbdv.200900112
Rezai T, Bock JE, Zhou MV, Kalyanaraman C, Lokey RS, Jacobson MP (2006) Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: successful in silico prediction of the relative permeabilities of cyclic peptides. J Am Chem Soc 128(43):14073–14080. doi:10.1021/ja063076p
Xiang T, Xu Y, Anderson BD (1998) The barrier domain for solute permeation varies with lipid bilayer phase structure. J Membr Biol 165(1):77–90
Kalyanaraman C, Jacobson MP (2007) An atomistic model of passive membrane permeability: application to a series of FDA approved drugs. J Comput Aided Mol Des 21(12):675–679. doi:10.1007/s10822-007-9141-z
QikProp, version 3.2, Schrodinger, LLC, New York, NY (2009)
Roux B, Simonson T (1999) Implicit solvent models. Biophys Chem 78(1–2):1–20
Bashford D, Case DA (2000) Generalized born models of macromolecular solvation effects. Annu Rev Phys Chem 51:129–152. doi:10.1146/annurev.physchem.51.1.129
Chang CE, Gilson MK (2004) Free energy, entropy, and induced fit in host-guest recognition: calculations with the second-generation mining minima algorithm. J Am Chem Soc 126(40):13156–13164. doi:10.1021/ja047115d
Chang CE, Chen W, Gilson MK (2007) Ligand configurational entropy and protein binding. Proc Natl Acad Sci USA 104(5):1534–1539. doi:10.1073/pnas.0610494104
Head MS, Given JA, Gilson MK (1997) “Mining Minima”: direct computation of conformational free energy. J Phys Chem A 101:1609–1618
Kolossvary I (1997) Evaluation of the molecular configuration integral in all degrees of freedom for the direct calculation of binding free energies: application to the enantioselective binding of amino acid derivatives to synthetic host molecules. J Am Chem Soc 119:10233–10234
Potter MJ, Gilson MK (2002) Coordinate systems and the calculation of molecular properties. J Phys Chem A 106:563–566
Fujikawa M, Nakao K, Shimizu R, Akamatsu M (2007) QSAR study on permeability of hydrophobic compounds with artificial membranes. Biorganic Med Chem 15:3756–3767
MacroModel, version 9.8, Schrodinger, LLC, New York, NY (2010)
Maestro, version 9.0, Schrodinger, LLC, New York, NY (2009)
Caves LS, Evanseck JD, Karplus M (1998) Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci 7(3):649–666. doi:10.1002/pro.5560070314
Monticelli L, Sorin EJ, Tieleman DP, Pande VS, Colombo G (2008) Molecular simulation of multistate peptide dynamics: a comparison between microsecond timescale sampling and multiple shorter trajectories. J Comput Chem 29:1699–1865
MATLAB version 7.11.0. Natick, The MathWorks Inc., Massachusetts (2010)
Grochowski P, Trylska J (2008) Continuum molecular electrostatics, salt effects, and counterion binding—a review of the Poisson-Boltzmann theory and its modifications. Biopolymers 89(2):93–113. doi:10.1002/bip.20877
Acknowledgments
This work is supported by NSF RAC CHE060073 and in part by the National Institutes of Health through the NIH Director’s New Innovator Award Program 1-DP2-OD007237 to R.E.A. R.V.S. would like to thank Dr. Ozlem Demir, Dr. Luke Czapla, Adam Van Wart, Brian R. Atwood, and Dr. Sara E. Nichols for useful discussion.
Supporting Information Available
Supporting information includes conformational search convergence plots.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Swift, R.V., Amaro, R.E. Modeling the pharmacodynamics of passive membrane permeability. J Comput Aided Mol Des 25, 1007–1017 (2011). https://doi.org/10.1007/s10822-011-9480-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-011-9480-7